Paper Reading : Fast, scalable generation of high-quality protein multiple sequence alignments us
0.简介

摘要中说道目前大多数都是使用渐进式路线启发式算法计算,但是对于成千上万个序列数据集时,这些方法可能已经到达瓶颈。难以保证在很大数量级数据上保证质量。本文介绍的clustal omega的新程序,它可以快速地对齐几乎任何数量的蛋白质序列,并提供精确的对齐。测试效果:在较小的测试用例中,包的精度与高质量的对齐器的精度相似。在更大的数据集上,clustal-omega在执行时间和质量方面优于其他包。Clustal Omega还具有强大的功能,可以向现有路线中添加序列并利用信息,从而使VastamountofFP在公共数据库(如PFAM)中重新计算信息。
1.介绍
对于长度为L的N个序列,精确计算N个序列之间最优对齐的方法具有 O(LN) [L的N次方]的计算复杂度,这使得即使是很小数量的序列也无法实现。大多数自动方法都是基于“渐进对齐”启发式(Hogeweg和Hesper,1984),它按照“导向树”中的分支顺序,对较大的子对齐中的序列进行对齐。这种方法的复杂度约为O(N2),通常可以使几千个中等长度的序列,但是很难使比对比这个大得多。渐进式方法是一种“贪婪算法”,在初始对准阶段所犯的错误在以后无法纠正。为了抵消这种影响,制定了一致性原则(Notredame等人,2000年)。这使得可以生产新一代更精确的对准器(例如T-Coffee(Notredame等人,2000))但是
以计算简便为代价。这些方法给出了5-10%更准确的对齐,如在基准上测量,但被限定为几百个序列。
在本报告中,我们介绍了一个名为clustal omega的新程序,它可以精确地对齐几乎任何大小的产品。我们已经用它在几个小时内在一个处理器上生成超过190000个序列的比对。在基准测试中,它明显比最广泛使用的、快速的方法更精确,并且在精度上与一些密集的慢速方法相当。它还具有强大的功能,允许用户重用其对齐,以避免在每次新序列可用时重新计算整个对齐。
逐步对准逼近标度的关键是用于生成指导树的方法。正常情况下,这需要将所有的N序列相互对齐,以满足 O(N2)的时间和内存要求。具有45万个序列的蛋白质家族正在出现,并将在各种大规模的基因组测序项目中变得普遍。目前,唯一一种能够对超过10000个序列进行常规比对的方法是mafft/parttree(katoh和toh,2007)。它速度很快,但会导致精度损失,必须通过迭代和其他启发式方法进行补偿。对于Clustal Omega,我们使用MBED的改进版本(Blackshields等人,2010年),其复杂性为O(N log n),并生成与传统方法一样精确的导向树。MBED的工作原理是将每个序列“嵌入”到一个N维的空间中,其中N与对数N成正比。然后用一个N元素向量替换每个序列,其中每个元素只是到N个“参考序列”。这些序列可以很快地聚类通过标准方法,如k-means或upgma。在clustal-omega中,然后使用非常精确的HHalign 包(so¨ding,2005)计算对齐,该包对齐了两个复杂的隐马尔可夫模型(eddy,1998)。
Clustal Omega具有许多功能,可以向现有路线添加序列,或使用现有路线帮助对齐新序列。一个创新是允许用户指定一个profile HMM,该HMM来自于与输入集同源的序列的对齐。然后将序列与这些“外部文件”对齐,以帮助它们与输入集的其余部分对齐。已经有很多可用的HMM集合,来自许多资源,例如 Pfam (Finn et al, 2009) ,现在这些可以用来帮助用户对齐他们的序列。 这里是否可以考虑将对齐比较好的序列先进行HMM,再使用profile HMM对其余部分对齐。
3.结果
3.1序列对齐的准确性
精度测量多序列对齐算法准确性的标准方法是使用参考对准的基准测试集,参照三维结构生成。在这里,我们展示了在三个基准上测试的一系列软件包的结果:BAliBASE (Thompson et al, 2005), Prefab (Edgar, 2004) and an extended version of HomFam (Blackshields et al, 2010)。对于这些测试,我们只使用所有程序的默认设置来报告结果,但有两个例外,这两个例外是为了允许 MUSCLE (Edgar, 2004) 和 MAFFT 调整HomFam中最大的测试用例。对于大于3000个序列的测试用例,我们使用–maxiter参数设置为2运行MUSCLE ,以便在合理的时间内完成对齐。其次,我们运行了几个不同的MAFFT 包程序。MAFFT (Katoh et al, 2002) 由一系列程序组成,这些程序可以单独运行,也可以通过设置了–auto标志自动调用脚本。当序列的数量和长度很小时,这个flag选择运行一个缓慢的、基于一致性的程序(L-INS-i)。当数字超过内置阈值时,使用传统的渐进式校准器(FFT-NS-2)。后者也是默认情况下运行的程序,如果调用MAFFT 时没有设置flags 。对于非常大的数据集,必须在命令行上设置–part tree flag,然后使用非常快速的导向树计算。

BAliBASE 基准测试的结果如表I所示。BAliBASE 分为六个“参考值”。每个参考值都给出了平均分数,以及总运行时间和平均总列(TC)分数,这些分数给出了恢复的总对齐列的比例。1.0分表示与基准完全一致。mafft包有两行:MAFFT(auto)和MAFFT default。在大多数(203/218个)BAliBASE 测试用例中,序列的数量很小,脚本运行L-INS-i,这是一个使用一致性启发式 (Notredame et al, 2000) 的低精度程序, 它也用在MSA probs (Liu et al, 2010), Probalign, Probcons (Do et al, 2005) 和 T-Coffee程序里。这些程序都只限于少量的序列,但往往给出精确的对齐。这明显反映在表一中的时间和平均得分上。这些包的时间范围从25分钟到22小时,准确度从55%到61%的栏正确。Clustal Omega同样的运行只需9分钟,但其准确度与Probcons和T-Coffee相似。
表的其余部分主要由使用渐进对齐的程序执行。其中一些速度非常快,但与基于一致性的程序和Clustal Omega相比,这种速度的精确度有了相当大的下降。这里最弱的项目是Clustal W (Larkin et al, 2007) ,其次是PRANK (Lo ¨ytynoja and Goldman, 2008)。PRANK的目的不是为了distantly related sequences,而是为了给系统发育工作提供良好的对齐,同时特别注意间隙。这些间隙位置不包括在这些试验中,因为它们往往在结构上不守恒。Dialign (Morgenstern et al, 1998) 不使用一致性或渐进对齐,而是基于查找最佳局部多重对齐。FSA (Bradley et al, 2009)使用成对排列和“序列退火”‘sequence annealing’的抽样,并且在过去已经被证明提供了良好的核苷酸序列排列。

The Prefab benchmark test results见表二。在这里,根据序列的一致性百分比将结果分为五组。总分情况大致在53%到73%。基于一致性的程序MSA probs,MAFFTL-INS-i,Probalign,Probconsand T-Coffee,是最精确但运行时间较长的程序。Clustal Omega接近于一致性程序的不精确性,但速度要快得多。然后,与MUSCLE, MAFFT, Kalign (Lassmann and Sonnhammer, 2005) 和 Clustal W的快速渐进式程序存在差距。

使用HomFam测试多达50000个序列的大型校准结果如表3所示。这里,每一个序列都由至少五个序列的基于 Homstrad (Mizuguchi et al, 1998)结构的序列的核心组成,然后将这些序列插入来自相应的同源pfam域的序列的测试集中(a test set of sequences from the corresponding, homologous, Pfam domain.)。这提供了要对齐的非常大的序列集,但测试仅在具有已知结构的序列上执行。只有一些程序能够提供这种大小的数据集的支持,我们将比较限制在 Clustal Omega, MAFFT, MUSCLE and Kalign。具有默认设置的MAFFT 有20000个序列的限制,我们只在表iii的最后一部分使用带有–part tree的MAFFT 。当获得超过3000个序列时,MUSCLE 会变得越来越慢。因此,对于大于3000个序列,我们使用了更快但不太准确的–maxiters 2设置的MUSCLE ,它将迭代次数限制为2次。总的来说,Clustal Omega很容易成为表iii中最精确的程序。运行时显示,MAFFT default和kalgin在较小的测试用例中运行得非常快,而MAFFT –part tree在最大的家族中运行得非常快。然而,随着序列数量的增加,Clustal Omega 表现确实很好。我们还有两个大于5万序列的测试用例,但是不可能从MUSCLE 或kalgin得到结果。这些也在补充信息中描述。
表三给出了用HomFam评估的四个程序的总运行时间。图1逐个解决了这些运行时。对于小家族来说,Kalign 速度很快,但不具有可伸缩性,总体来说,MAFFT 在所有测试用例大小上都比其他程序快,但Clustal Omega的伸缩性(最好?)相似。图1中的点表示具有不同平均序列长度和成对一致性的不同族。因此,可伸缩性趋势是模糊的,较大的点通常出现在较小的点之上。图三显示了可伸缩性数据,其中长度逐渐增加的子集仅从一个大家族中采样。这减少了成对一致度和序列长度的可变性。

图1Clustal Omega (red), MAFFT (blue), MUSCLE (green) and Kalign (purple) 与HomFam测试集序列数的比对时间。平均序列长度由点大小呈现。两个轴都有对数运算刻度。Clustal Omega 和Kalign在整个比较范围内都是以默认的速度进行的。MUSCLE用–maxiters 2进行 N>3000 个序列。MAFFT 使用–parttree运行N>10000个序列。
3.2 EPA
External profile alignment Clustal Omega可以从a profile HMM中读取来自先前存在的对齐的额外信息。例如,如果用户
希望对齐一组globin序列并已经拥有对齐的globin序列,此对齐可以转换为profile HMM并与序列输入文件一起使用。这个HMM在这里被称为“外部profile”,并以这种方式被称为“外部profile alignment”(epa)。在EPA过程中,输入集中的每个序列都与外部文件对齐。然后将来自外部文件的伪计数信息逐位置传输到输入序列。理想情况下,这将与特定蛋白质或感兴趣的领域(如在宏基因组项目中使用的领域)的大型精选比对一起使用。每次发现新的序列时,应仔细维护并将其用作EPA的外部文件,而不是将输入序列从头开始对齐。Clustal Omega还可以使用传统对齐方法将序列与现有对齐对齐。用户可以将序列逐个添加到路线或将一组对齐的序列与路线对齐。
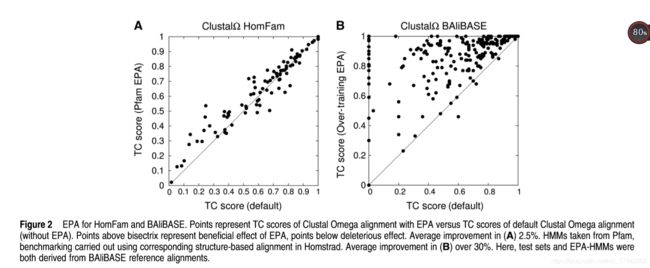
在本文中,我们用两个例子来说明EPA方法。首先,我们从上一节中获取94个 HomFam测试用例,并使用对应的 Pfam HMM 来实现EPA。在EPA之前,测试用例的平均准确度是正确对齐的Homstrad 位置的0.627,但是在EPA之后,它上升到0.653。这是图2A中测试用例的测试用例图。每个点是一个测试用例,使用EPA将Clustal Omega的TC分数与该分数相对应。第二个例子如图2b所示。在这里,我们取所有的BAliBASE 参考集,并使用Clustal Omega将它们正常对齐,得到正确对齐的0.554列的基准结果,如表1所示。对于EPA,我们使用基准参考对齐它们自身作为外部设施。结果现在跳到0.857列正确。这是一个超过30%的跳跃,虽然它不是一个有效的测量方法,与其他程序比较, Clustal Omega准确度,但它确实说明了EPA在外部比对中使用信息的潜在能力。
3.3迭代
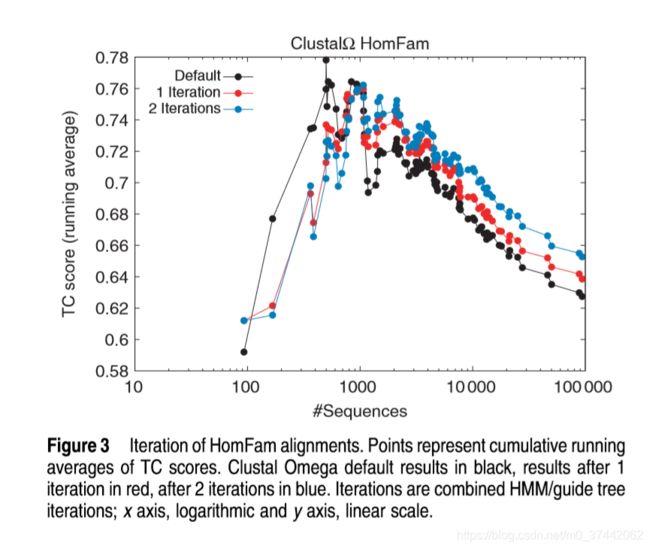
EPA也可用于简单的迭代方案中。一旦从一组输入序列生成MSA,就可以将其转换为HMM,并用于EPA 来帮助重新调整输入序列。这也可以与完全重新计算导向树相结合。在图3中,我们展示了HomFam中每个测试用例的一次和两次迭代的结果。该图被绘制为所有测试用例的运行平均TC分数,其中N使用对数刻度在水平轴上绘制。对于一些较小的测试用例,迭代实际上会产生有害的影响。然而,一旦你得到了近1000个或更多的序列,就会出现一个明显的趋势。你拥有的序列越多,迭代的效果就越有利。对于更大的测试用例,应用两次迭代将变得越来越有利。这一结果证实了EPA作为一项总体战略的有效性。它也证实了在对齐大量序列时的困难,但给出了一个部分解决方案。它还提供了一个非常简单但有效的迭代方案,不仅用于指导树迭代(如许多包中所使用的),而且用于路线本身的迭代。
4.讨论
自20世纪80年代中期以来,MSA方法的主要突破是逐步对齐和一致性的使用。另外,最近的研究关注的是基准测试集的速度或准确度。速度的提高是显著的,但是,除了两个主要的例外,这些方法基本上仍然是O(N2) ,并且不能扩展到大于1万个序列的数据集。这里使用的两个例外是这里使用的mBed和MAFFT Part Tree,Part Tree速度更快,但要牺牲准确性,至少从这里的基准判断是这样的。第二组最新进展涉及准确性。这往往侧重于基准测试的结果,这是一个潜在的有争议的问题 (Aniba et al, 2010; Edgar, 2010)。我们现有的基准测试集范围有限,并且严重偏向于单域球状蛋白。这有可能导致方法在基准上表现良好,但在实际情况中不那么灵活或有用。提高准确性的一个发展是招募额外的同源物来大量增加输入数据集。这对于基于一致性的方法和小数据集似乎很有效。然而,似乎在没有进一步发展的情况下,以这种方式获得的额外精度是有限度的。额外的序列也可能带来噪声,并大大增加了计算问题的复杂性。这可以部分通过迭代来确定,但是,EPA对高质量的参考比对可能是一个更好的解决方案。这也增加了对可视化这种大型路线的方法的需求,以便检测问题。
第二个发展重点是使用外部信息,如RNA结构 (Wilm et al, 2008) 或蛋白质结构预测(Pirovano et al, 2008)。EPA是一种新的方法,它允许用户利用他们自己的或公开可用的对齐方式中的信息。它不会强制新序列完全遵循旧的对齐方式。新的序列通过渐进排列相互对齐,但外部文件中的信息有助于提供序列中每个位置最有可能出现哪些氨基酸的信息。大多数方法试图从蛋白质进化的一般模型中预测这一点,并以二级结构预测为基础。在本文中,我们已经证明,即使使用来自Pfam 的批量生产的校准作为外部配置,对于一个大型通用测试用例集的准确性也会有小幅度的提高。这为用户提供了一组新的可能性,可以利用大型的、公开可用的对齐中包含的信息,并鼓励数据库提供商提供高质量的对齐。Clustal X 取得巨大成功的原因之一是非常友好的图形用户界面 (GUI)。然而,由于基于web的服务的广泛可用性(gui由基于web的前端服务器提供),这并不像过去那样重要。此外,还有一些非常高质量的对齐查看器和编辑器,如Jalview (Clampetal,2004)和Seaview(Gouyetal,2010),它们读取 Clustal Omega输出或可以直接调用 Clustal Omega。
5.材料和方法
Clustal Omega的材料和方法是根据GNU Lesser通用公共许可证授权的。源代码以及Linux、FreeBSD、Windows和Mac(Intel和PowerPC)的预编译二进制文件可在http://www.clustal.org上找到。Clustal Omega仅作为命令行程序提供,它使用GNU样式的命令行选项,还接受ClustalW样式的命令选项,以便向后兼容并轻松集成到现有管道中。Clustal Omega是用C和C++编写的,它使用了许多优秀的免费软件包。我们使用了
Sean Eddy的Squid库(http://selab.janelia.org/software.html)用于Sequence I/O,允许使用多种文件格式。我们使用David Arthur的K-Means++代码(Arthur和Vassilvitskii,2007)快速聚类序列向量。fast UPGMA和导向树处理程序的代码来自MUSCLE (Edgar, 2004).。我们使用OpenMP 库来实现成对距离和对齐匹配状态的多线程计算。Clustal Omega的API文档是源代码的一部分,此外还可以从http://www.clustal.org/omega/clustalo-api/获得。所有算法的详细信息见随附的补充信息。使用的基准是BAliBASE 3 (Thompson et al, 2005), PREFAB 4.0 (posted March 2005) (Edgar, 2010)和使用来自PFAM(版本25)和Homstrad(截至2011-06-13)的序列的新构建的数据集(Homfam)(Mizuguchi等人,1998年)。
比较的程序可以从以下网址获得:
ClustalW2, v2.1 (http://www.clustal.org)
DIALIGN 2.2.1 (http://dialign.gobics.de/)
FSA 1.15.5 (http://sourceforge.net/projects/fsa/)
Kalign 2.04 (http://msa.sbc.su.se/cgi-bin/msa.cgi)
MAFFT6.857(http://mafft.cbrc.jp/alignment/software/source.html)
MSAProbs 0.9.4 (http://sourceforge.net/projects/msaprobs/files/)
MUSCLE version 3.8.31 posted 1 May 2010 (http://www.drive5. com/muscle/downloads.htm)
PRANKv.100802,2August2010(http://www.ebi.ac.uk/goldman-srv/ prank/src/prank/)
Probalign v1.4 (http://cs.njit.edu/usman/probalign/)
PROBCONSversion1.12(http://probcons.stanford.edu/download.html)
T-Coffee Version 8.99 (http://www.tcoffee.org/Projects_home_ page/t_coffee_home_page.html#DOWNLOAD).补充信息
补充信息可在分子系统生物学网站(www.nature.com/msb)上获得。