零代码复现-TCGA联合GEO免疫基因结合代谢基因生信套路(二)
前面的分析中,下载TCGA和GEO的数据,并进行简单的处理,接下来就是相关基因集的获取和整理,为后期聚类和降维做准备工作(数据准备:https://blog.csdn.net/weixin_43949246/article/details/134215103)
三、获取免疫基因和代谢相关的基因集
这里可以选择从GSEA官网下载免疫和代谢相关基因,或者换成其他的基因也是可以的
1、取对数,TCGA的数据一般跨度比较大,所以在使用limma做差异之前,需要取一个log进行后续的分析(http://www.sxdyc.com/logarithmicdata)

运行完成后,点击下载即可

2、直接提取相关基因的表达谱矩阵
准备好基因集后,可以收到一个一个捕获基因的表达谱,也可以使用我们的工具
首先获取这两个基因集合并为all.gene.txt

使用工具,提取特定的基因集(提取部分基因表达,http://www.sxdyc.com/geneExpression)
需要上传两个文件,一个是全基因的表达谱数据,一个是基因的列表信息
运行完成后,直接下载数据

四、TCGA数据差异分析
1、准备分组文件
接下来进行差异分析,需要准备一个分组文件,这里选择 4.特征基因表达谱的准备 文件夹中生成的dat.select.txt 的文件


从14号位置往后数两个字符,其中01为肿瘤组织,11为正常组织

02其实也是肿瘤组织,只是很多分析中会去掉,因为01是原发性肿瘤,保存一下(所以只保留了01和11的样本)

2、差异分析
这里其实有两种做法的,一种是拿免疫+代谢相关基因表达谱进行差异分析,一种是拿全部的基因做差异分析,筛选出差异基因后,在和免疫+代谢相关基因集取交集。(这里选择的是免疫+代谢相关基因表达谱进行差异分析)
http://www.sxdyc.com/diffLimmaAnalyse

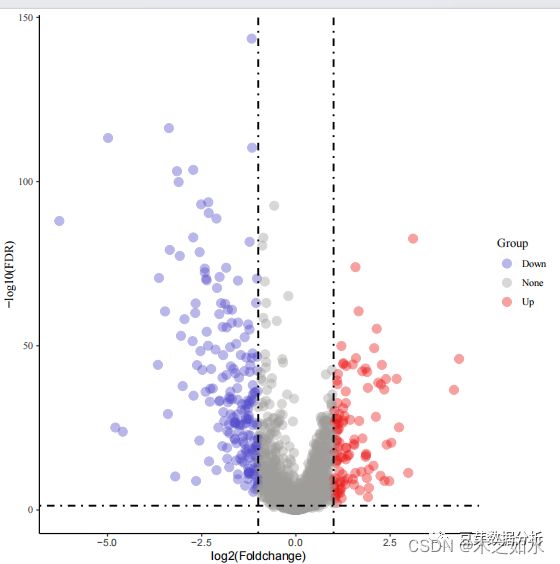
3、差异分析的火山图的绘制
从limma差异分析中下载差异分析的结果
获取绘制火山图的数据,包含三列,顺序分别为基因,差异倍数,p值

http://www.sxdyc.com/visualsVolcano

补充一点,如果这里用全部的基因做差异,需单独使用venn图的工具取一个交集
4、差异基因表达的热图
差异基因的获取,用excel的筛选工具,对volcan0.txt进行筛选(标准:logFC选择大于1或者小于-1,FDR<0.05,当然这里的标准可以进行修改),把相关基因提取放在heatmap.gene.txt文件中
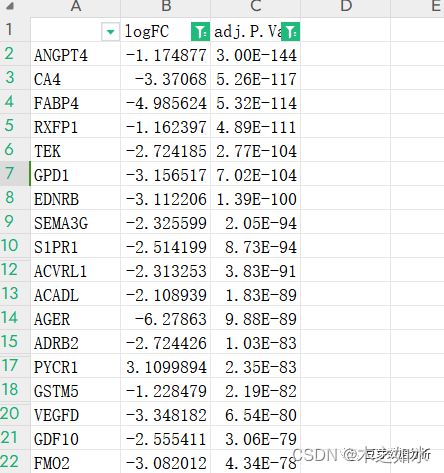

先通过工具获取差异基因的表达谱(http://www.sxdyc.com/geneExpression)
运行成功后,下载数据,并改名为heatmap.exp.txt

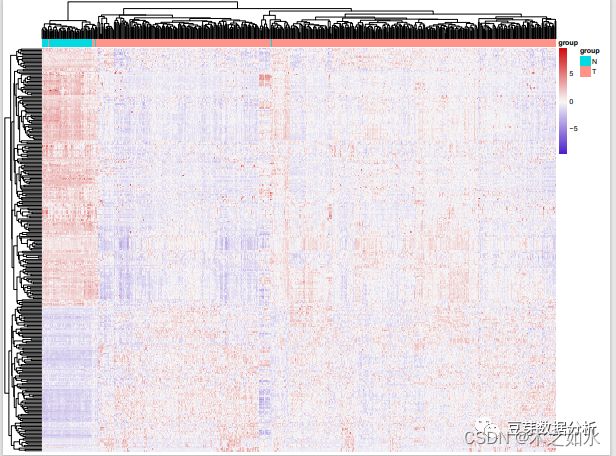
在使用工具绘制热图(http://www.sxdyc.com/visualsClusterHeat)

这里需要注意的是
行注释为:基因的分组信息,如上调,下调的分组,代谢还是免疫基因的分组等等;
列注释为:样本的分组信息,例如这个癌组织和癌旁组织
热图的颜色:从低到高的颜色
是否显示行名:这里需要显示基因名,就选yes
是否显示列名:这里需要显示样本命,就选yes
是否进行行聚类:如果需要对基因进行聚类,就选yes
是否进行列聚类:如果需要对样本进行聚类,就选yes
图片的高度和宽度设置好
提交后等待运行成功即可下载

五、TCGA和GEO差异基因获取和预后数据的整理
1、差异基因集的获取,获取TCGA和GEO数据集的共有基因
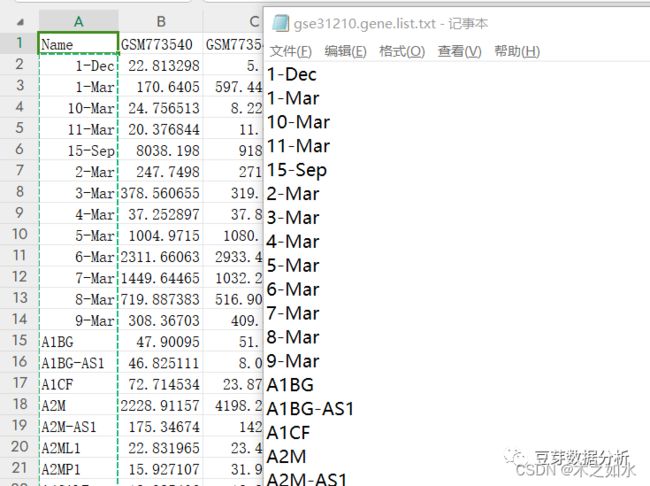
首先,需要将GEO的数据中的基因名单独复制到gse31210.gene.list.txt文件中
使用venn/upset图绘制工具,获取交集基因(http://www.sxdyc.com/visualsVennUpset)
准备绘制venn图的文件,如这里只要两个组
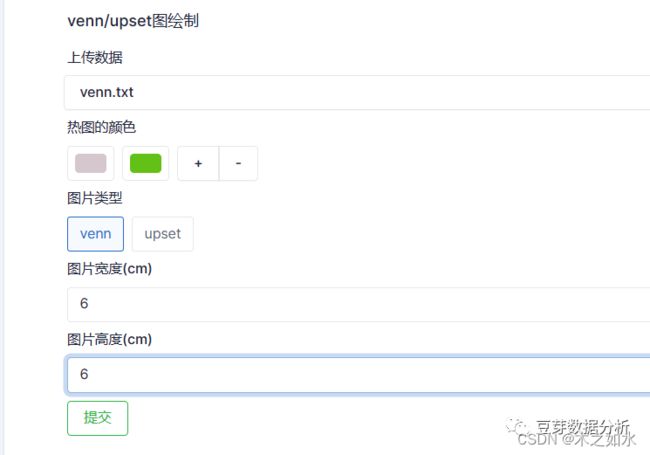
上传后,绘制venn图,这里的颜色指的是分组的颜色(工具写错了,后面会改掉),设置图片的宽度和高度,运行完成后,直接下载即可


这时候就需要提取共有基因表达谱,包括TCGA和GEO的表达谱数据,为后面的分型和建模做准备。
2、TCGA数据集肿瘤组织生存数据的准备和表达谱矩阵的准备
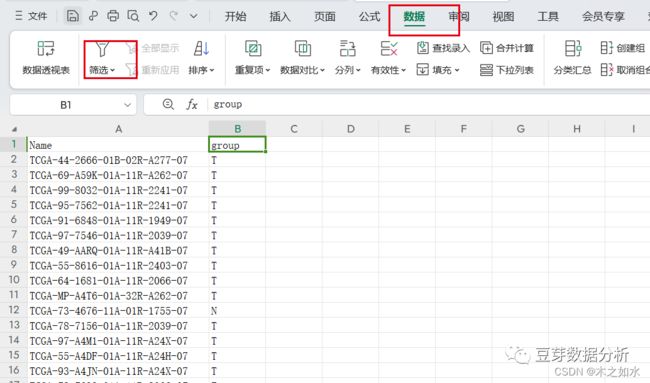
提取TCGA的肿瘤组织的表达谱,在5.差异基因热图获取的group.txt和heatmap.exp.txt,打开group.txt,数据筛选,删掉N的样本,只有肿瘤组织的样本,另存为group1.txt(只保留第一列的样本名,去掉列名)。

打开生存数据TCGA.merge.cli.txt,去掉生存时间为0的样本

准备两列数据,第一列数据为生存时间大于0的样本,第二列为肿瘤组织的样本,用excel选择1-12位字符串,接下來复制C列的数据,选择性粘贴在C列,选择数值(这一步很重要)

然后把A列的数据复制在C列之下,用excel筛选共有的样本

开始-条件格式-突出显示单元格规则-重复值(选择C列之后)
将没有颜色的行删掉

这时候将B列的样本名复制为group1.txt的文件中。
同时将C列中重复的样本复制在生存数据中,将不存在的样本删掉TCGA.merge.cli.txt,一样的方法去掉多余的样本

打开heatmap.exp.txt文件,转置,如下所示,改为heatmap.exp1.txt

使用提取部分基因表达工具提取肿瘤组织的表达谱(http://www.sxdyc.com/geneExpression),当然也可以手动一个一个删,这样比较麻烦

运行完成后,直接下载,并改名为tcga.T.dat.txt
用excel打开tcga.T.dat.txt文件,然后,提取样本的1-12位字符串
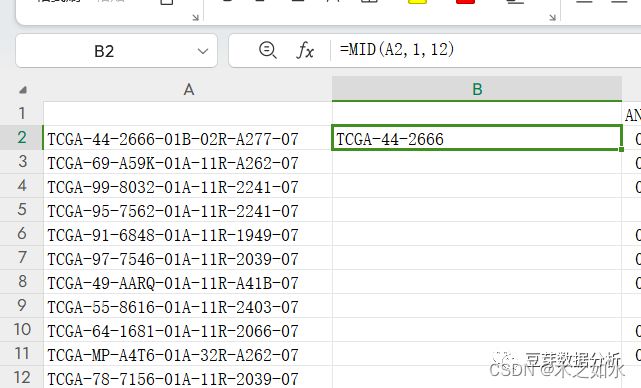
接下來复制B列的数据,选择性粘贴在A列,选择数值(这一步很重要)
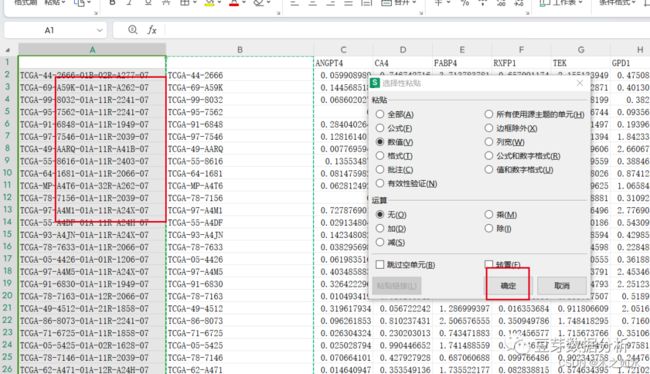
然后删除B列的信息,只留A列的信息即可
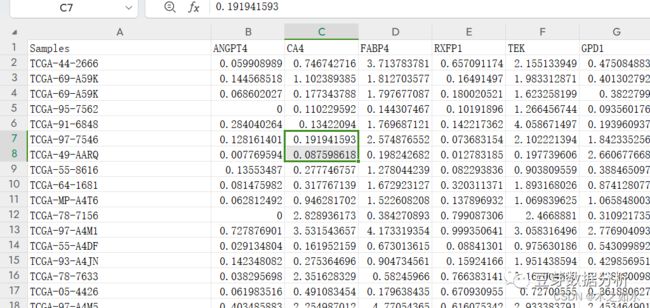
该文件保存一下即可
3、GEO数据生存时间的筛选和表达谱矩阵的获取
首先获取共有基因表达谱,准备文件(2.geo.pre\GSE31210\gse31210.dat.process.txt),6.TCGA和GEO差异基因获取和预后数据的整理中获取的(com.set.txt),使用工具(http://www.sxdyc.com/geneExpression)提取gse31210数据集表达谱
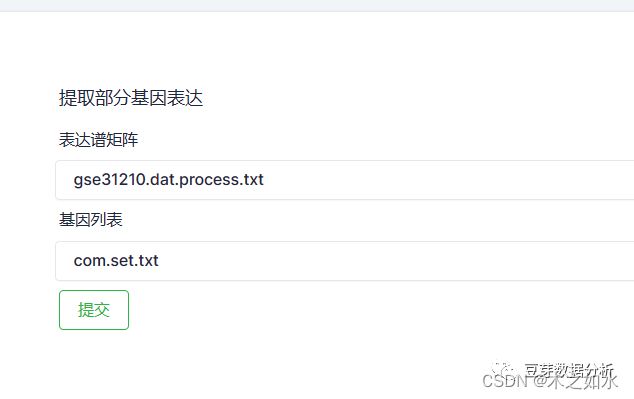
运行完成后,下载即可

下载后改名为gse31210.select.gene.txt
打开gse31210的生存数据,删掉正常组织,保留我们想要的临床信息

修改生存时间名字(习惯性修改为OS.time和OS),并将生存状态Ailve替换为0,dead替换为1
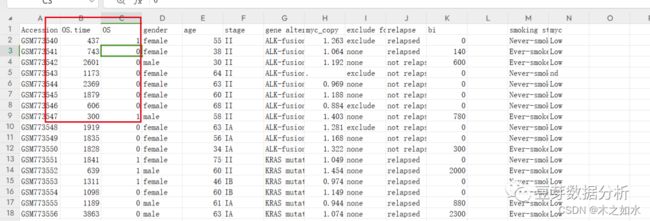
并将样本复制出现,保存在gse.sample.txt

打开gse31210.select.gene.txt文件,在excel转置一下,提取肿瘤组织的表达谱数据,和TCGA数据一样
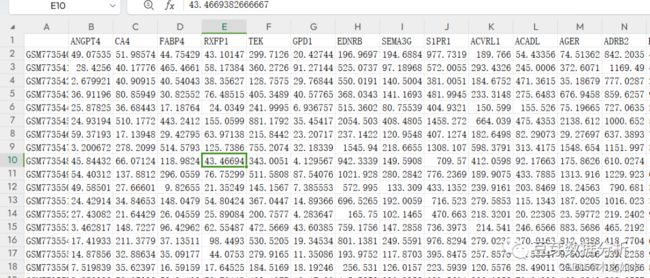

运行完成后,下载并改名为gse31210.T.data.txt
接下来就是分子亚型和风险模型的构建
#################################
到这里关键基因集的获取和生存数据准备基本就结束,如果有兴趣可以关注公众号(豆芽数据分析)
有零代码复现相关问题,可以付费进入零代码答疑群和大家一起交流,群费为9.9元,拒绝白嫖党(添加微信:ShenxinBS003或者ShenxinBS001)