【学习笔记】山东大学生物信息学-02 序列比较
课程地址:山东大学生物信息学
文章目录
- 二、序列比较
-
- 2.1 认识序列
- 2.2 序列相似性
- 2.3 替换记分矩阵
- 2.4 序列两两比较:打点法
- 2.5 序列两两比较:序列比对法(定量)
- 2.6 一致性和相似度
- 2.7 在线双序列比对工具
- 2.8 BLAST 搜索
- 2.9 多序列比对介绍
- 2.10 在线多序列比对工具
- 2.11 多序列比对的编辑和发布
- 2.12 寻找保守区域
二、序列比较
2.1 认识序列
sequence 就是个字符串 string。
FASTA 格式:
第一行:大于号加名称或其他注释
第二行以后:每行 60 个字母(也有 80 的,不一定)
2.2 序列相似性
-
相似的序列 → 相似的结构 → 相似的功能
-
可预测未知结构和功能的蛋白质的结构和功能
-
序列一致度与相似度:
◆ 一致度(identity):如果两个序列长度相同,那么它们的一致度定义为它们对应位置上相同的残基的数目占总长度的百分比。
◆ 相似度(similarity):如果两个序列长度相同,那么它们的相似度定义为它们对应位置上相似的残基与相同的残基的数目和占总长度的百分比。
残基两两相似的量化关系被替换记分矩阵所定义。
2.3 替换记分矩阵
● 替换记分矩阵(substitution matrix):反映残基之间相互替换率的矩阵,它描述了残基两两相似的量化关系。分为 DNA 替换记分矩阵和蛋白质替换记分矩阵。
★ 3 种常见 DNA 序列的替换记分矩阵
- 等价矩阵(unitary matrix):最简单的替换记分矩阵,其中,相同核苷酸之间的匹配得分为
1,不同核苷酸间的替换得分为0。由于不含有碱基的理化信息和不区别对待不同的替换,在实际的序列比较中较少使用。 - 转换-颠换矩阵(transition-transversion matrix):核酸的碱基按照环结构特征被划分为两类,一类是嘌呤(A/G),有两个环;另一类是嘧啶(C/T),只有一个环。如果 DNA 碱基的替换保持环数不变,则为转换;如果环数发生变化,则成为颠换。在进化过程中,转换发生的频率远比颠换高。为了反映这一情况,通常该矩阵中转换的得分为
-1,而颠换的得分为-5。 - BLAST 矩阵:经过大量实际对比发现,如果使被比对的两个核苷酸相同时得分为
+5,反之为-4,则比对效果较好。这个矩阵广泛地被 DNA 序列比较所采用。

★ 3 种常见蛋白质序列的替换记分矩阵
-
等价矩阵(unitary matrix):与 DNA 等价矩阵道理相同,相同氨基酸之间的匹配得分为 1。不同氨基酸间的替换得分为 0。在实际的序列比对中较少使用。
-
PAM 矩阵(Dayhoff 突变数据矩阵):PAM 矩阵基于进化原理。如果两种氨基酸替换频繁,说明自然界易接受这种替换,那么这对氨基酸替换得分就应该高。PAM 矩阵是目前蛋白质序列比较中最广泛使用记分方法之一,基础的 PAM-1 矩阵反映的是进化产生的每一百个氨基酸平均发生一个突变的量值(统计方法得到)。PAM-1 自乘 n 次,可以得到 PAM-n,即发生了更多次突变。(根据要比较的序列之间亲缘关系远近来选择适合的 PAM 矩阵,如果亲缘关系远,即有很多突变,n 就越大,反之 n 越小。)
◆ PAM-250 矩阵:对角线上的数值为匹配氨基酸的得分;其他位置上,≥0 的得分代表对应氨基酸对为相似氨基酸。

-
BLOSUM 矩阵(blocks substitution matrix):BLOSUM 矩阵是通过关系较远的序列来获得矩阵元素的。PAM-1 矩阵是基于相似度较高(>85%)的序列比对计算产生的,那些进化距离较远的矩阵,如 PAM-250 是通过 PAM-1 自乘得到的。即,BLOSUM 矩阵的相似度是根据真实数据产生的,而 PAM 矩阵是通过矩阵自乘外推来的。和 PAM 矩阵一样,BLOSUM 矩阵也有不同编号,如
BLOSUM-80代表该矩阵是由一致度≥80% 的序列计算而来。同理,BLOSUM-62指该矩阵由 一致度≥62% 的序列计算而来。
◆ BLOSUM-62:对角线上的数值为匹配氨基酸的得分;其他位置上,≥0 的得分代表对应氨基酸对为相似氨基酸。

Q1: 选 PAM-1 还是 PAM-250?

Q2: 选 PAM-? 还是 BLOSUM-?

- 对于关系较远的序列之间的比较,由于 PAM-250 是推算而来,所以其准确度受到一定限制,BLOSUM-45 更具优势。
- 对于关系较近的序列之间的比较,用 PAM 或 BLOSUM 矩阵做出的比对结果,差别不大。
- 最常用的:BLOSUM-62
★ 其他 2 种蛋白质序列比对的替换记分矩阵
-
遗传密码矩阵 (genetic code matrix,
GCM) : 遗传密码矩阵通过计算一个氨基酸转换成另一个氨基酸所需的密码子变化的数目而得到,矩阵的值对应为据此付出的代价。
◆ 如果变化一个碱基就可以使一个氨基酸的密码子转换为另一个氨基酸的密码子,则这两个氨基酸的替换代价为1;
◆ 如果需要 2 个碱基的改变,则替换代价为2;
◆ 再比如从 Met 到 Tyr 三个密码子都要变,则代价为3。
◆ 遗传密码矩阵常用于进化距离的计算,其优点是计算结果可以直接用于绘制进化树,但是它在蛋白质序列比对(尤其是相似程度很低的蛋白质序列比对)中,很少被使用。
-
疏水矩阵: 根据氨基酸残基替换前后疏水性的变化而得到得分矩阵。若一次氨基酸替换疏水特性不发生太大的变化,则这种替换得分高,否则替换得分低。
2.4 序列两两比较:打点法
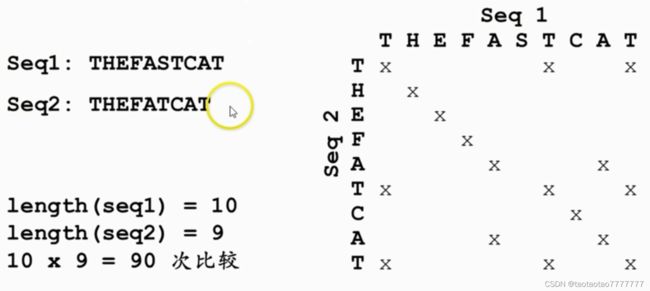
- 打点法:相同的打点。
- 连续的对角线、对角线的平行线,代表两条序列中相同的区域。

- 可以用一条序列自己对自己打点,从而可以发现序列中的重复片段。这样的打点矩阵必然是对称的,并且有一条主对角线。在横向或纵向上,与主对角线平行的短平行线所对应的序列片段就是重复的部分;包括主对角线在内的平行线出现的次数就是重复次数。

- 发现串联重复序列 (
tandem repeat):
如 Seq1: FASABCABCABCTHE
◆ 重复次数:在半个对角线内,包括主对角线在内的所有等距平行线的个数。
◆ 重复单元:最短的平行线对应的序列。
◆ 短串联重复序列 (short tandem repeat,STR) 也叫做微卫星 DNA, 是一类广泛存在于真核生物基因组中的 DNA 串联重复序列。它由 2-6bp 的核心序列组成,重复次数通常在 15-30 次。STR 具有高度多态性,即存在重复次数的个体间差异,而且这种差异在基因遗传过程中一般遵循孟德尔共显性遗传规律,所以它被广泛用于法医学个体识别、亲子鉴定等领域。

- Dotlet 在线打点工具:Dotlet 需要安装 java。
详见视频:序列两两比较:打点法-02 P34

2.5 序列两两比较:序列比对法(定量)
- 序列比对 (
alignment),也叫对位排列、联配、对齐等。运用特定的算法找出两个或多个序列之间产生最大相似度得分的空格插入和序列排列方案。 - 序列 s 和 t 的比对:把 s 和 t 这两个字符串上下排列起来,在某些位置插入空格(空位,
gap),然后依次比较它们在每一个位置上字符的匹配情况,从而找出使这两条序列产生最大相似度得分的排列方式和空格插入方式。

双序列比对及算法
- 全局比对(globalalignment):用于比较两个长度近似的序列。Needlernan-Wunsch 算法,1970 年,SaulNeedleman 和 Christian Wunsch 两人首先将动态规划算法应用于两条序列的全局比对,这个算法后称为 Needleman-Wunsch 算法。
详见视频:序列两两比较:序列比对法-02 全局比对 P37


- 局部比对(local alignment):用于比较一长一短两条序列。1981 年 Temple Smith 和 Michael Waterman 对局部比对进行了研究,产生了 Smith-Waterman 算法。
详见视频:序列两两比较:序列比对法-03 局部比对 P38


2.6 一致性和相似度
- 如果两个序列长度相同:
一致度 (identity) = (一致字符的个数 / 全局比对长度) × 100%
相似度 (similarity) = (一致及相似的字符的个数 / 全局比对长度) × 100%

- 如果两个序列长度不相同:
一致度 (identity) = (一致字符的个数 / 全局比对长度) × 100%
相似度 (similarity) = (一致及相似的字符的个数 / 全局比对长度) × 100%

- 无论两个序列长度是否相同,都要先做双序列全局比对,然后根据比对结果及比对长度计算它们的一致度和相似度。
2.7 在线双序列比对工具
EMBL 全局双序列比对工具
-
详见视频:在线双序列比对工具-01 P40
详见视频:在线双序列比对工具-02 Gap 的类型及分值设置 P41 -
EMBL → Global Alignment → Needle → 输入/上传要比对的 2 条序列
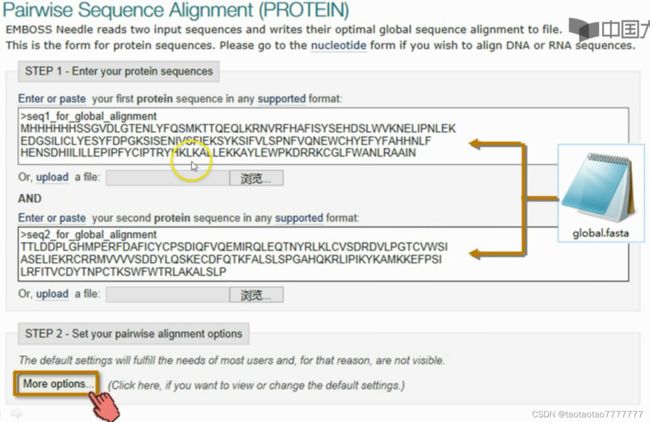
-
参数设置 More options:
- MATRIX:默认选 BLOSUM-62,或按亲缘关系选择。
- GAP OPEN:出现第一个空位时的罚分值,默认比 GAP EXTEND 罚分多。
- GAP EXTEND:出现连续多个空位时的罚分值(除第一个空位外),默认罚分比 GAP OPEN 少。
- GAP OPEN 比 GAP EXTEND 罚分大时,空位集中,开第一个空位缺口代价大,但鼓励出现连续空位。
案例:已知两条序列绝大部分相似,其中一条序列的功能区中另一条序列中缺失,需要通过序列比对,找出这个缺失的功能区,选择集中的空位。 - GAP OPEN 比 GAP EXTEND 罚分小时,空位分散,连续空位代价大,鼓励出现短空位。
案例:比对同源序列,已知两条序列很相似,结构功能差不多,选择分散的空位。 - 如果对结果没有预期,保持默认参数即可。
- END GAP PENALTY:GAP 结尾的罚分,默认 false 即可。


EMBL 局部双序列比对工具
- 详见视频:在线双序列比对工具-03 P42
- EMBL → Local Alignment → Water → 输入/上传要比对的 2 条序列 → Submit
- 序列 1 两头对不上(红色)的部分,在比对结果中直接被忽略;
序列 2 结尾比对补上的部分,也直接被忽略
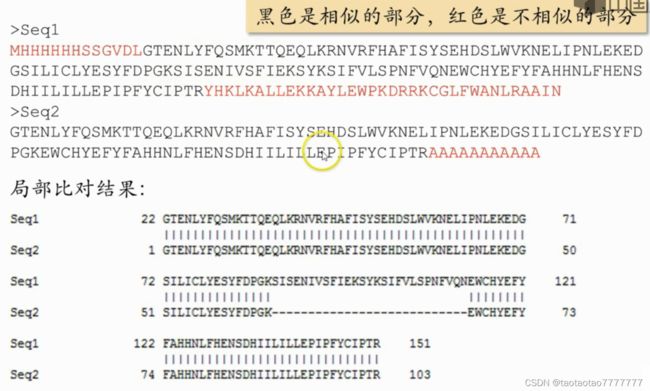
- 全局比对与局部比对比较:
- 其他在线双序列比对工具
| 软件名 | 比对类型 |
|---|---|
| EMBL | Global/Local |
| PIR | Global |
| Lalign | Global/Local |
| LAGAN | Global |
| AlignMe | Alignment of Membrane Proteins |
| MCALIGN | Alignment of non-coding DNA sequences |
| Biotools | Global/Local |
2.8 BLAST 搜索
- BLAST (Basic Local Alignment Search Tool) 基本局部比对搜索工具,是目前最常用的数据库搜索程序。
- BLAST 的要点是片段对。所谓片段对是指两个给定序列中的一对子序列,它们的长度相等,且可形成无空位的完全匹配。
- BLAST 基本原理:BLAST 首先找出探测序列和目标序列间所有的匹配程度超过一定阈值的序列片段对,然后对片段对根据给定的相似性阈值进行延伸,得到一定长度的相似性片段,最后给出高分值片段对 (high-scoring pairs, HSPs)。改进后的 BLAST 允许空位的插入。

BLAST 的种类
- BLAST 实际上是综合在一起的一组工具的统称,它不仅可用于直接对蛋白质序列数据库和核酸序列数据库进行搜索,而且可以将带搜索的核酸序列翻译成蛋白质序列后再进行搜索,或反之,以提高搜索效率。

Blastp: 用蛋白质序列搜索蛋白质序列数据库(常用)Blastn: 用核酸序列搜索核酸序列数据库(常用)Blastx: 将核酸序列按 6 条链翻译成蛋白质序列后搜索蛋白质序列数据库tblastn:用蛋白质序列搜索核酸序列数据库,数据库中的核酸序列要按 6 条链翻译成蛋白质序列后再搜索。tblastx: 将核酸序列按 6 条链翻译成蛋白质序列后搜索核酸序列数据库,数据库中的核酸序列要按 6 条链翻译成的蛋白质序列后再搜索。(针对新发现的序列)- 根据搜索算法分:
标准 BLAST,PSI-BLAST,PHI-BLAST等。
标准 BLAST
- 详见视频:BLAST 搜索-03 实操 P46



- BLAST 结果:
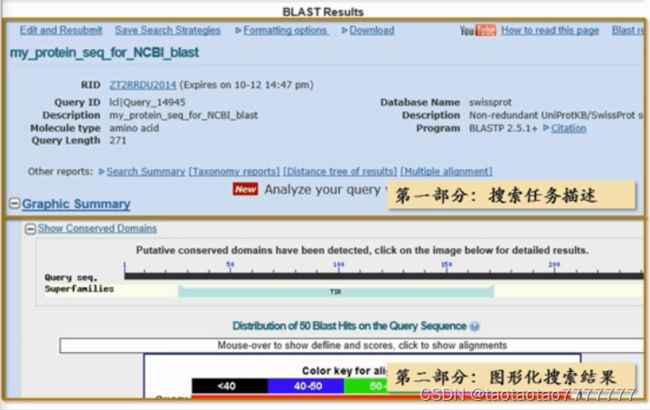

Total score(匹配得分) 和Query cover(覆盖度) 分别决定匹配序列的颜色和长短。

E value(期望值):E 值越接近零,说明输入序列与当前这条序列为同一条序列的可能性越大。- 匹配结果根据 E 值由小到大排序,随着 E 值增大,
Total score成反比逐渐降低,但Ident一致度与 E 值并非完全成反比(因为 BLAST 为了提高速度,没有做双序列比对,牺牲了一定准确度。表中的一致度是 BLAST 搜索完成后,针对搜索到的 50 条序列做双序列比对后得到的)。
PSI-BLAST (撒大网搜索)
- 有时基本 BLAST 搜索还是不能满足需要。比如,你想通过一条蛋白质序列,搜罗出一个庞大的蛋白质家族。 如果运行基本的 BLAST 搜素,你只能找到那些和探索序列十分相近的序列,而其他那些远源序列就找不到了。
PSI-BLAST(Position-Specific Iterated BLAST, 位点特异性迭代 BLAST)
PSI-BLAST 的特色是每次用位置特异权重矩阵 (Position-Specific Scoring Matrix,PSSM) 搜索数据库后再利用搜索的结果重新构建 PSSM,然后用新的 PSSM 再次搜索数据库,如此反复 (iteration) 直至没有新的结果产生为止。(找到朋友的朋友)- 详见视频:BLAST 搜索-04 PSI BLAST P47
- 第一轮搜索结果和标准 BLAST 一样。

- 点 Go 进行第二轮搜索(可以指定列出搜索结果的前多少列)


PHI-BLAST (精准搜索)
-
详见视频:BLAST 搜索-05 PHI-BLAST P48
-
PHI-BLAST(Pattern-Hit Initiated BLAST, 模式识别 BLAST): 能找到与输入序列相似的并符合某种特定模式 (pattern) 的序列。 -
例如,N-糖基化位点基序 (N-glycosylation site motif) 总是符合以下特定模式:以 Asn(N) 开始,然后紧跟除了 Pro§ 之外的任何一个氨基酸,再紧跟 Ser(S) 或者 Thr(T), 再紧跟除 Pro 外的任何一个氨基酸。
- 利用正则表达式搜索:N{P}[ST]{P}
- 用正则表达式书写的符合模式:{L}GEx [GAS] [LIVM]x(3,7)
{}代表匹配除大括号内以外的任意内容(除…以外)[]代表匹配中括号中的任意内容(其中之一)x代表任意字符x(3,7)代表 3-7 个 x 字符- 例如: VGEAAMPRI 符合 VGEAAYPRI 不符合
-
这种序列特征模式可能代表某个翻译后修饰的发生位点,也可以代表一个酶的活性位点,或者一个蛋白质家族的结构域、功能域。

-
PHI-BLAST 和 PSI-BLAST 可以联合使用

其他 BLAST
- 详见视频:BLAST 搜索-06 其他 BLAST P49

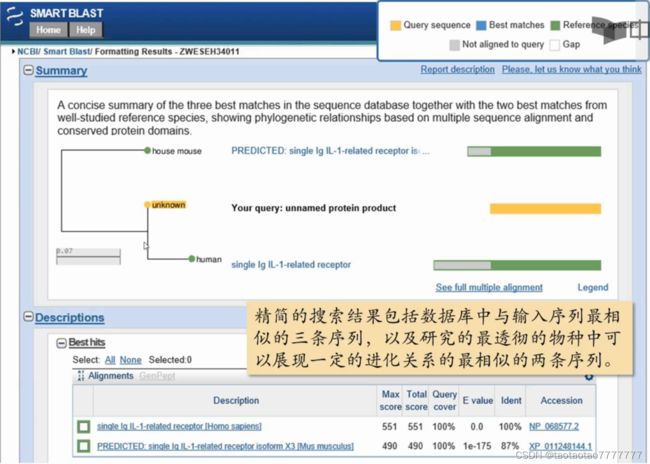
- SmartBLAST:精简的搜索结果包括数据库中与输入序列最相似的三条序列,以及研究的最透彻的物种中可以展现一定的进化关系的最相似的两条序列。

- 互联网上的免费搜索工具(利用时差选择不同的 BLAST 工具)
| 位置 | 服务器 | 网址链接 |
|---|---|---|
| USA | NCBI | http://www.ncbi.nlm.nih.gov/BLAST |
| Europe | ExPASy | http://web.expasy.org/blast |
| Europe | Uniprot | http://www.uniprot.org/blast/ |
| Japan | DDBJ | http://blast.ddbj.nig.ac.jp |
WU-BLASTWU 代表 Washington University。比 NCBI-BLAST 更灵敏,在插入空位的算法上更灵活。- Smith and Waterman (
SSEARCH): 有点儿慢,但是比 BLAST 更准确。 FASTA: 有点儿慢,但是对于 DNA 序列的比较比 BLAST 更准确。BLAT: 用于小的序列(如 cDNA 等)在大基因组中的搜索。
2.9 多序列比对介绍
多序列比对-用途及算法
-
多序列比对 (multiplealignment),对两条以上的生物序列进行全局比对。
-
多序列比对的主要用途:
- 确认:一个未知的序列是否属于某个家族。
- 建立:系统发生树,查看物种间或者序列间的关系。
- 模式识别:一些特别保守的序列片段往往对应重要的功能区域,通过多序列比对,可以找到这些保守片段。
- 已知推未知:把已知有特殊功能的序列片段通过多序列比对做成模型,然后根据该模型推测未知的序列片段是否也具有该功能。
- 其他:预测蛋白质/RNA 二级结构等。
-
多序列比对的算法:目前所有的多序列比对工具都不是完美的,它们都使用一种近似的算法。(通过多序列比对看趋势,大体位置,牺牲准确度)

-
多序列比对注意事项:
- 太多的序列受不了。一般 10-15 条序列,最好别超过 50 条。
- 关系太远的序列受不了。两两之间序列相似度低于 30% 的一组序列,作多序列比对会有麻烦。
- 关系太近的序列受不了。两两之间序列相似度大于 90% 的序列,有再多条都等于只有一条。
- 短序列受不了。多序列比对支持一组差不多长的序列,个别很短的序列属于捣乱分子。
- 有重复域的序列受不了。如果序列里包含重复域,大多数多序列比对的程序都会出错,甚至崩溃。
-
序列的名字有几点建议:
- 名字里不要有“空格”,用 “_” 代替 “空格”。
- 不要用特殊字符,(比如中文,@,#,&,^等)。
- 名字的长度不要超过 15 个字符。
- 一组序列里,不要有重名的序列。
- 如果不按上述几点建议命名的话,多序列比对的工具会在不告知你的情况下修改你的序列名称。
2.10 在线多序列比对工具
-
Clustal 最常用的多序列比对工具
-
TCOFFEE 最新的多序列比对工具之一
-
MUSCLE 最快的多序列比对工具之一
-
部分提供多序列比对在线使用的网站
| 网站名称 | 服务器位置 | 网址链接 |
|---|---|---|
| EBI | Clustal-Omega | http://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Expasy | Clustal W | http://www.ch.embnet.org/software/ClustalW.html |
| Sf-Clustal | Clustal O/W2 | http://www.clustal.org/ (仅下载) |
| EBI | Tcoffee | http://www.ebi.ac.uk/Tools/msa/toffee |
| TCC FFEE | Tcoffee | http://www.fofee.org/ |
| EBI | Muscle | http://www.ebi.ac.uk/Tools/msa/muscle/ |
| MUSCLE | Muscle | http://www.drive5.com/muscle/ (仅下载) |
EMBL
-
详见视频:在线多序列比对工具-01 EMBL P52
-
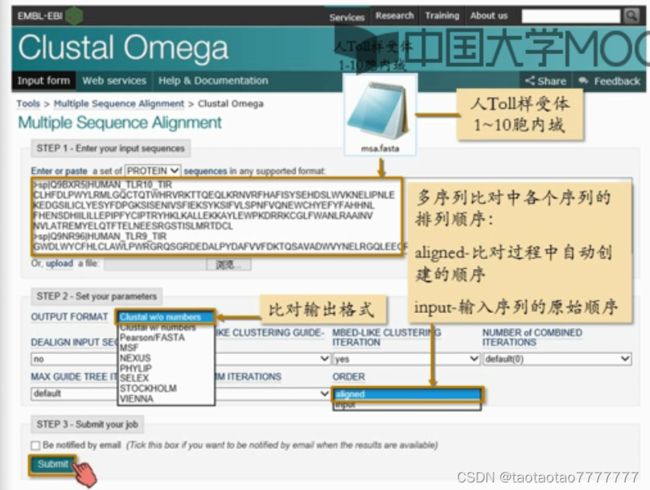
ORDER
aligned 比对过程中自动创建的顺序
input 按输入序列的原始顺序输出结果
-
Download Alignment File

-
Show Colors
Red:疏水的(红色)
Blue: 酸性的(蓝色)
Magenta: 碱性的(品红)
Green: 羟基+胺+碱性(绿色)
Gray: 其他(灰色)

-
每行比对结果最后都有星星点点的标记,标记密集的区域也就是这些序列间的保守区域。
符号 含义 * 完全保守的一列,即,这一列的残基完全相同。 : 这一列的残基有大致相似的分子大小及相同亲疏水性,即这一列残基或相同或相似。 · 在进化过程中,残基的分子大小及亲疏水性被一定程度上保留了,但是有替换发生在不相似的残基间。(有相似的也有不相似的) (空白) 完全不保守的一列(完全不相似)。

Result Summary

Phylogenetic Tree注意:这个不是真正的系统发生树。

- 要得到系统发生树,在
Alignments中将比对结果发送给专门做系统发生树的软件Send to ClustalW2_Phylogeny。

Tcoffee
-
详见视频:在线多序列比对工具-02 Tcoffee P53
-
Tcoffee http://tcoffee.crg.cat
多序列比对工具,算法上与 Clustal 系列类似,准确度上比 Clustal 系列略高,但计算耗时也比 Clustal 系列略高。 -
许多网站都提供 Tcoffee 的在线使用
SIB http://tcoffee.vital-it.ch
EBI http://www.ebi.ac.uk/Tools/msa/tcoffee
CNRS http://www.igs.cnrs-mrs.fr/Tcoffec/tcoffee_cgi/index.cgi
Max-Planck http://toolkit.tucebingen.mpg.dc/t_coffec
CBSU http://cbsuapps.tc.cornell.edu/t_coffec.aspx
EMBnet http://www.es.embnet.org/Scrvices/MolBio/t-coffee -
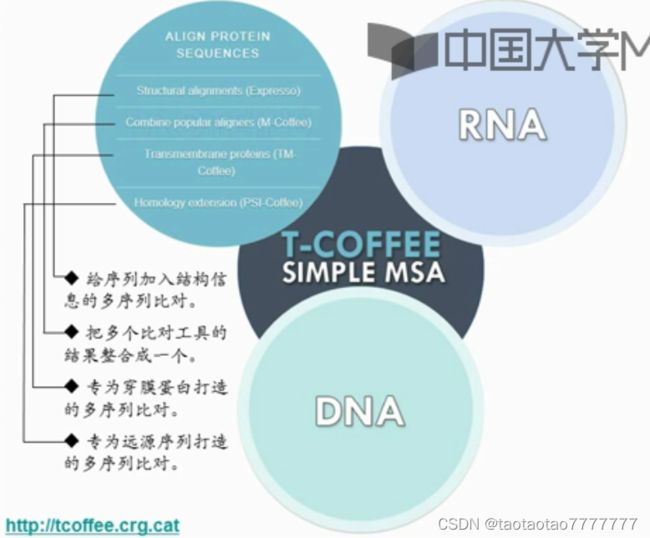
Protein
Structural alignments (Expresso) :给序列加入结构信息的多序列比对。
Combine popular aligners (M-Coffee):把多个比对工具的结果整合成一个。
Transmembrane proteins (PSI/TM-Coffee):专为穿膜蛋白打造的多序列比对。
Homology extension (PSI-Coffee):专为远源序列打造的多序列比对。
-
Expresso:给序列加入结构信息的多序列比对。
提供的信息越少,比对时间越长,有必要留个邮箱等结果。
比对结果的颜色代表比对结果的好坏,红黄绿蓝,逐渐变差。



多序列比对的保存格式
- 详见视频:在线多序列比对工具-03 多序列比对的保存格式 P54
- 多种保存格式:网页格式、clustal 格式(多序列比对)、fasta 格式(写完一条再写下一条)、phylip 格式(建树用)
- 如果比对工具输出的格式里没有我想要的哪种,可以通过第三方软件进行格式转换,比如:
fmtseq: http://evol.mcmaster.ca/Pise/5.a/fmtseq.html
http://www.bioinformatics.org/JaMBW/1/2
2.11 多序列比对的编辑和发布
- 为了能对多序列比对的结果进行彩色显示和手工编辑,人们开发了多序列比对结果编辑器。
- Jalview 是一个特别常用的编辑器。http://www.jalview.org
- 详见视频:多序列比对的编辑和发布-01-02 Jalview P55-56
- 从 EMBL 多序列比对结果中快速启动 JalView。但是快速启动的 Jalview 功能不全!

- 下载到本地安装(需要 java)

- 导入多序列比对结果 clustal 文件
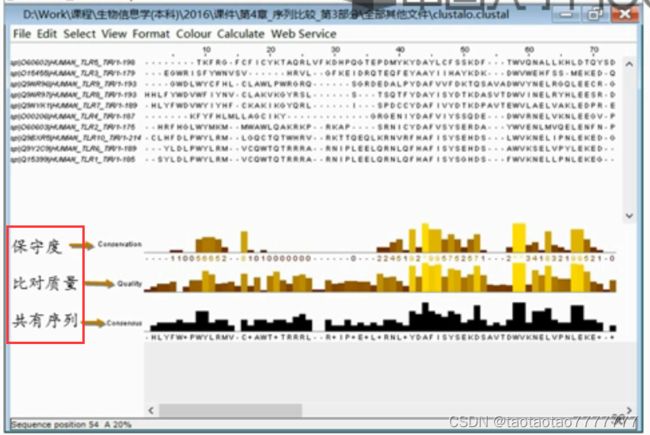
- Colour 上色
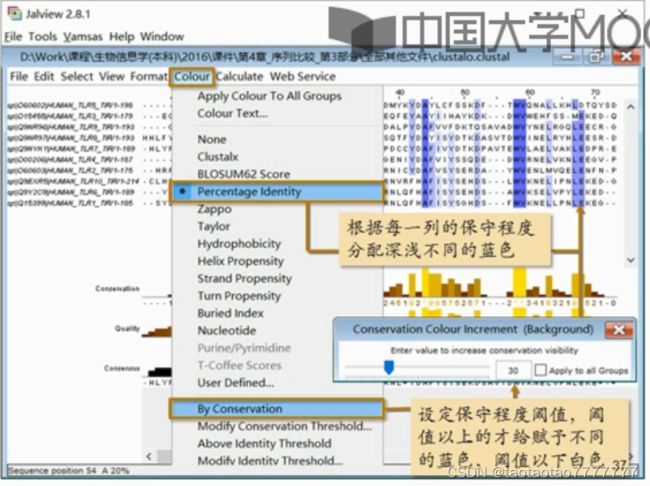
常见的 Clustal 系列配色方案

- 修整局部瑕疵:对局部手动调整

- 自动换行、设置字体

- 打开/关闭注释行
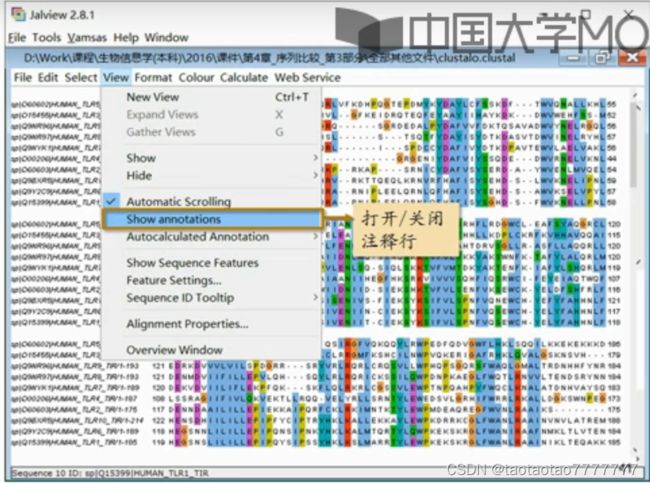
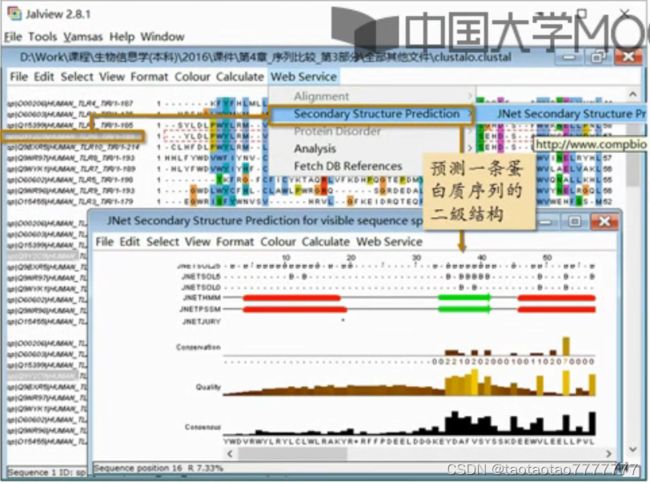
基本分析功能
- 详见视频:多序列比对的编辑和发布-02 Jalview P56
- 按照各种规则排序、为任意一对序列做双序列全局比对

- 为选中的一组序列创建系统发生树

- 预测一条蛋白质序列的二级结构
- 把序列比对保存成图片
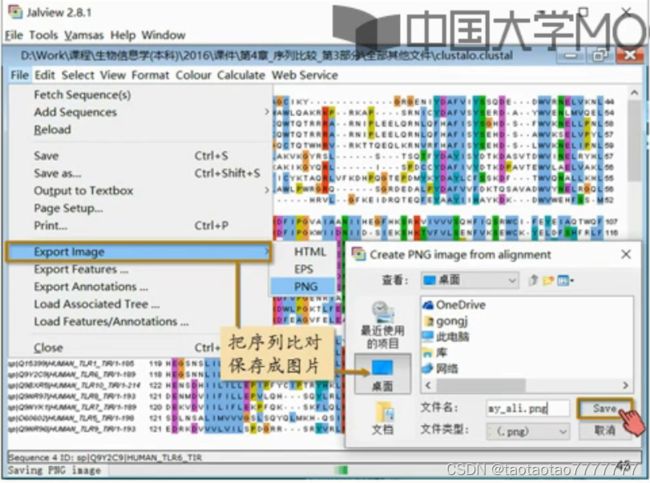
- 多序列比对美化工具
| 名称 | 网址 | 特点 |
|---|---|---|
| JalView | http://www.jalview.org | JAVA,可嵌入网页 |
| Boxshade | http://www.ch.embnet.org/software/BoX_form.html | 擅长黑白作图 |
| ESPript | http://lespript.ibcp.fr/ESPript/ESPript | 功能强大,很牛 |
| MView | http://bio-mview.sourceforge.net | 擅长转换成 HTML 源码 |
2.12 寻找保守区域
序列标识图
- 详见视频:寻找保守区域-01 P57
- 序列标识图 (sequence logo) 是以图形的方式依次绘出序列比对中各个位置上出现的残基。每个位置上残基的累积可反映出该位置上残基的一致性。每个残基对应图形字符的大小与残基在该位置上出现的频率成正比。但图形字符的大小并不等于频率百分比(否则每一列应该都是一样高的),而是经过简单统计计算后转化的结果。

- 图形字符的大小并不等于频率百分比,否则每一列字母总高度应该都是一样的,因为在字母高度的计算时涉及熵值,一列字母出现越混乱,熵值越大,字母越矮;越有规律,熵值越小,字母越高。
WebLogo 3
- 一款流行的创建序列标识图的软件:WebLogo 3 http://weblogo.threeplusone.com/
- 创建 WebLogo 例如输入多条启动子序列


序列基序:MEME
- 详见视频:寻找保守区域-02 MEME P58
- 在核酸/蛋白质序列中存在有特定模式 (pattern) 的序列片段,这些片段称为序列的基序 (
motif)。序列基序与生物功能密切相关。 - MEME 是一款可以自动从一组相关的 DNA 或蛋白质序列中发现序列基序的软件。http://meme-suite.org
- 上传原始序列,不需要提前做多序列比对

- 返回结果的各种格式


- 点击
more下的箭头↓可以看到放大的 sequence logo 获得具体的基序信息

- 右边的箭头
→可以提交基序给其他软件或数据库,针对该基序进行序列相似性搜索。

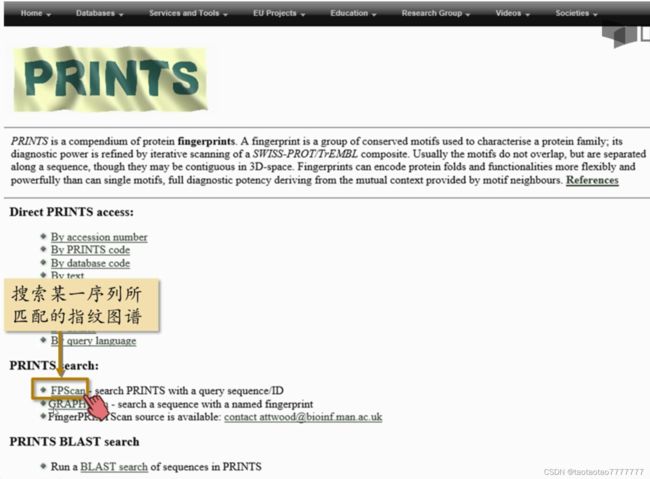
PRINTS 指纹图谱数据库
-
详见视频:寻找保守区域-03 PRINTS P59
-
一个蛋白质的指纹 (Prints)就是一组保守的序列基序,用于刻画蛋白质家族的特征。这些基序由多序列比对结果获得,且他们在氨基酸序列上是不相邻的,但是在三维结构中,他们可能紧密结合在一起。
-
PRINTS http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/ 是蛋白质序列指纹图谱数据库,存储了目前已发现的绝大多数蛋白质家族的指纹图谱。对于一个陌生的蛋白质,只要看看它的序列是否符合某个家族的图谱就可以对它进行分类并预测它的功能。
-
Direct PRINTS access: 查找的蛋白质指纹图谱有多种方式


-
TRANSFERRIN指纹图谱信息
-
View alignment查看创建指纹图谱所使用的多序列比对
-
View Structure以家族中某一蛋白质的结构为例,在线显示基序在三维结构中的位置
-
-
PRINTS searchFPScan指纹匹配:搜索某一序列所匹配的指纹图谱