富集分析原理和clusterProfiler包进行GO、KEGG富集分析详细说明
概念:
基因富集分析是指对于给定一组基因根据基因组注释信息(GO、KEGG)对基因进行聚类分析,即给定的基因是不是GO中的一个功能(或KEGG中的一个通路)。
基因的功能富集的目的是说明给定的基因集对哪些功能的影响有针对性的,不是随机影响的。
原理:
基因富集分析是通过研究给定的基因集在功能节点上是否过出现得到关注的基因集显著注释的功能节点。通常利用超几何分布等方法计算给定基因集在某个功能(或通路)上的P值,判断给定的基因集在功能(或通路)的基因数目超过了随机的期望,是一个小概率事件。

| 变量 | 差异表达分析 |
| N | 基因组所有基因、所有分析的基因 |
| x | 差异表达基因集中有功能F的基因 |
| M | N中具有某种功能(F)的基因总数 |
| K | 差异表达基因 |
clusterProfiler包进行GO、KEGG富集分析:
1.加载R包,下载R包请参考:
富集分析--R包--clusterProfiler下载安装与报错分析解决_Tian問的博客-CSDN博客
#加载包
library(clusterProfiler)
library(org.Hs.eg.db)
library(topGO)● clusterProfiler包由Y叔开发,可以进行基因及基因簇的分析和基因谱功能可视化,功能强大。
● org.Hs.eg.db人类的基因组注释包进行基因ID的转换
● topGO包辅助绘制GO富集分析结果的有向无环图
2、数据准备,筛选感兴趣的基因集
#自行选择筛选自己感兴趣的基因集
gene_set <- rownames(pro_result[which(pro_result$FDR < 0.01 & abs(pro_result$log2FC) >= 2),])● 根据自己的需求获取差异基因
3、基因ID转换
gene_symbol <- bitr(geneID = gene_set, #感兴趣的基因集
fromType="ENSEMBL", #输入ID的类型
toType=c("SYMBOL", "ENTREZID"), #输出ID的类型,可为多个
OrgDb="org.Hs.eg.db") #物种注释数据库● 可能出现部分ID无法匹配的结果,通常输出的ID少于输入的ID
4、GO富集分析
gene <- gene_symbol[,3]
CC <- enrichGO(gene = gene, #基因列表(转换的ID)
keyType = "ENTREZID", #指定的基因ID类型,默认为ENTREZID
OrgDb=org.Hs.eg.db, #物种对应的org包
ont = "CC", #CC细胞组件,MF分子功能,BP生物学过程
pvalueCutoff = 0.01, #p值阈值
pAdjustMethod = "fdr", #多重假设检验校正方式
minGSSize = 1, #注释的最小基因集,默认为10
maxGSSize = 500, #注释的最大基因集,默认为500
qvalueCutoff = 0.01, #q值阈值
readable = TRUE) #基因ID转换为基因名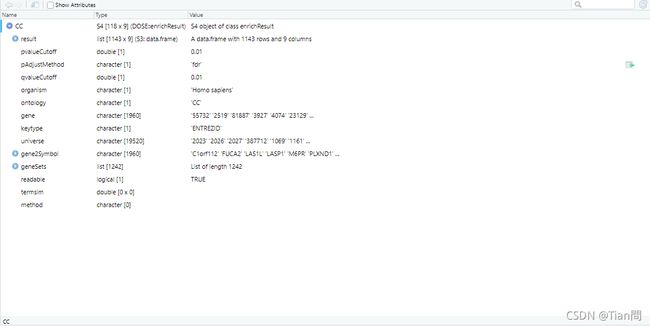
#展示GO的CC的富集结果result
df <- CC@result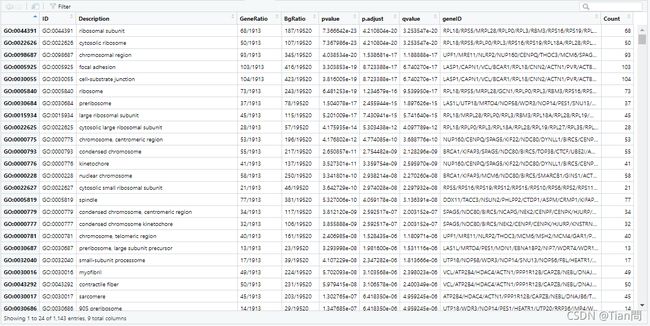
● ID:GO数据库ID
● Decription:基因功能描述
● GeneRAatio:K/x
● BgRatio:M/N
● pvalue,qvalue:p值和校正过的p值
● count:差异基因的数目
5、GO富集分析可视化
5.1绘制点图
dotplot(CC, #GO富集分析结果
x = "GeneRatio", #横坐标,默认GeneRation,也可以为Count
color = "p.adjust", #右纵坐标,默认p.adjust,也可以为pvalue和qvalue
showCategory = 20, #展示前20个点,默认为10个
size = NULL, #点的大小
title = "CC_dotplot" #设置图片的标题
)
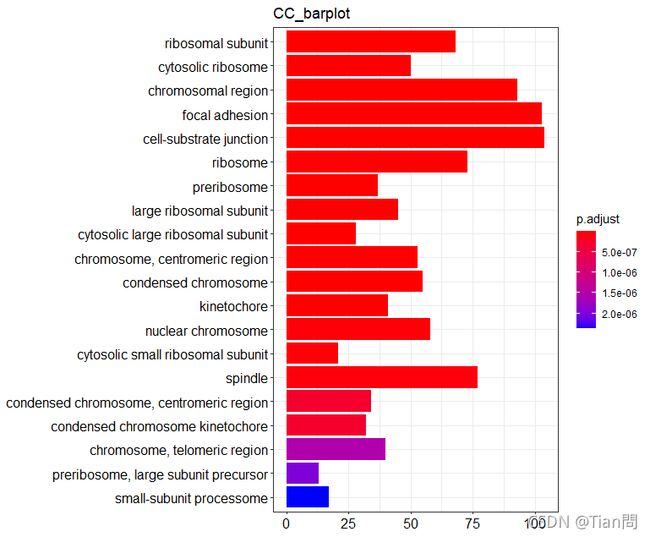
5.2绘制条状图
barplot(CC, #GO富集分析结果
x = "Count", #横坐标,默认Count,也可以为GeneRation
color = "p.adjust", #右纵坐标,默认p.adjust,也可以为pvalue和qvalue
showCategory = 20, #展示前20个,默认为10个
size = NULL,
title = "CC_barplot" #设置图片的标题
)
5.3GO富集分析的有向无环图(DAG)
plotGOgraph(CC, #输出enrichGO或gseGO的有向无环图(与输入的对象对应)
firstSigNodes = 10, #显著性节点的个数,默认10个
useInfo = "all",
sigForAll = T, #是否在所有节点展示score/p-value
useFullNames = T, #是否使用全称
)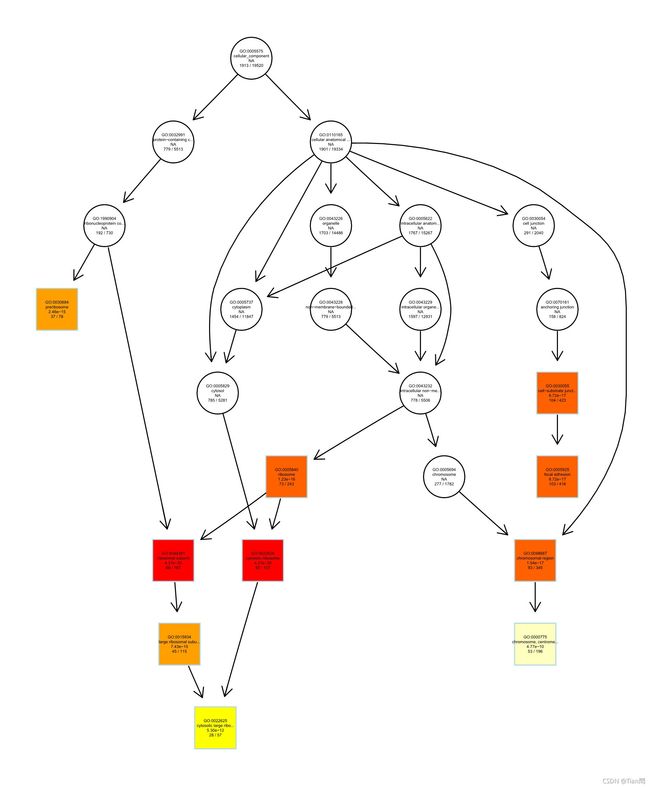
● 在GO富集分析的有向无环图(DAG)中,方形是默认输出的显著性最高的前10个节点;且颜色的深浅表示显著性,颜色越深,越显著。
● 图形中内容的含义:

自上而下,依次为:
● GO数据库中编号(ID)
● 节点功能注释
● p值
● K/M
6、KEGG富集分析
KEGG<- enrichKEGG(gene = gene, #基因列表(同GO)
organism = "hsa", #物种
keyType = "kegg", #指定的基因ID类型,默认为kegg
minGSSize = 1,
maxGSSize = 500,
pvalueCutoff = 0.01,
pAdjustMethod = "fdr",
qvalueCutoff = 0.01) 
#展示GO的CC的富集结果result
df <- KEGG@result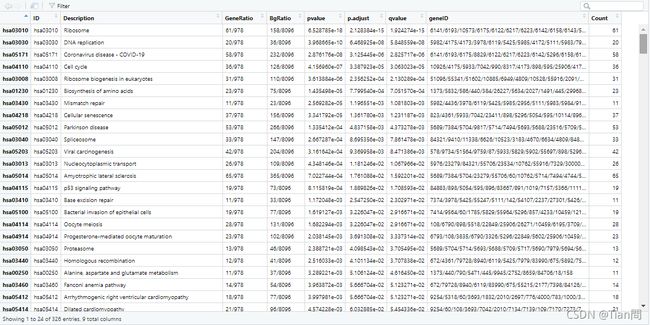
7、KEGG富集分析可视化
KEGG可视化的点图和条图和GO富集分析可视化一致,不在展示,且KEGG不可绘制DAG图
GO和KEGG富集分析还可以借助其他R包绘制诸如热图、弦图,大家可自行学习
感谢您的查看,致谢!(`・ω・´)ゞ(`・ω・´)ゞ
欢迎关注公众号《生信Tian問的笔记》ε≡٩(๑>₃<)۶ 一心向学
