Nat.Mach.Intell.|如何改进错义突变致病性预测?使用图注意神经网络试试
编译 | 杨慧丹
审稿 | 赵宸
本文介绍一篇来自哥伦比亚大学Yufeng Shen的研究团队最近发表在Nature Machine intelligence期刊上的一项研究。作者提出了一种基于图注意力神经网络的新方法来预测错义突变的致病性,即基于图的错义突变致病性预测器(graphical missense variant pathogenicity predictor , gMVP)。作者证明了gMVP可以改善对临床检测和基因研究中错义突变的理解。

研究背景与内容
错义突变是导致癌症遗传风险和发育障碍的主要因素,用于许多临床遗传诊断。然而,大多数罕见错义突变可能是良性的或只有极小的功能效应,临床遗传检测中报道的大多数罕见错义突变也意义不明确,导致模糊、混乱、过度治疗和错过临床干预时机。在人类基因研究中,人们利用罕见突变来识别新的风险基因,基于计算预选出具有破坏性的错义突变是提高其统计能力的必要步骤。因此,计算方法对于解释临床遗传学和疾病基因发现研究中的错义突变至关重要。
已有一些研究证明,错义突变的功能效应与三维蛋白质结构相关,汇集三维结构信息可以改善对功能效应的预测。该论文所提出的gMVP模型主要组成部分是一个图,具有捕获氨基酸预测特征的节点和协同进化强度加权的边,能够有效地汇集来自局部蛋白质上下文和功能相关的远端位置的信息。作者通过有监督学习与精心策划的致病性突变数据对模型进行训练与测试,研究证明了gMVP在识别TP53、PTEN、BRCA1和MSH2中的破坏性突变方面优于其他已发表的方法。此外,它实现了对神经发育障碍病例中de novo错义突变与对照组病例的最佳分离。最后,模型还支持通过迁移学习来优化对钠通道和钙通道的功能增益(gain-of-function,GOF)和功能损失(loss-of-function,LOF)预测。
研究方法
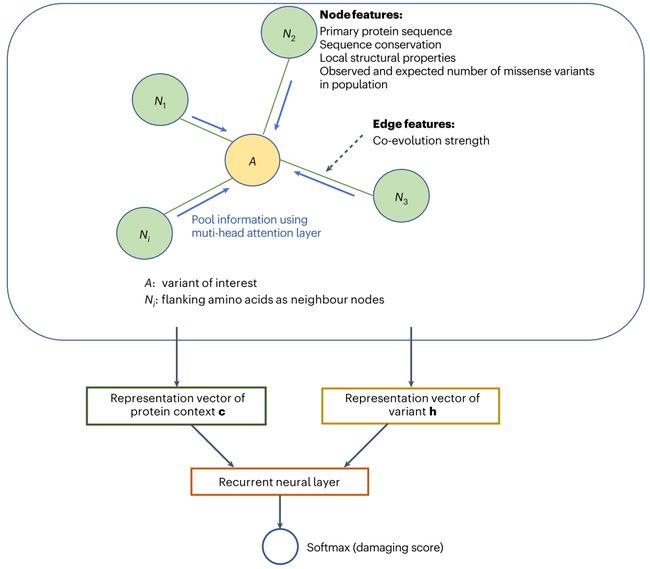
图1. gMVP模型概览
错义突变的功能效应取决于氨基酸替代的类型和蛋白质上下文。gMVP使用一个图来表示一个突变体及其蛋白质的上下文(上下文定义为128种侧翼氨基酸)。目标氨基酸是中心节点(橙色),侧翼氨基酸是上下文节点(绿色),所有上下文节点与中心节点相连,但彼此不相连。图中节点特征描述其序列保守性和局部结构性质,边的权重使用协同进化强度,可以有效汇集在远端但功能相关位置上的保守性和编码约束性信息。
作者使用一个图注意力神经网络来学习蛋白质序列和结构上下文的表示。其包括三个深度为1的密集层来将三个输入特征(中心节点x、上下文节点{ni}和边的特征向量{fi})分别编码为潜在表示向量h, {ti}和{ei},一个多头注意层来学习上下文的注意力权重(tanh(W[h, ti, ei])),然后得到上下文向量c。最后将向量c和h输入一个与softmax层相连接的递归神经层进行分类并生成预测得分。
研究结果
作者精心策划了人类群体中的致病性突变和随机的罕见错义突变数据来训练gMVP,并使用完全不同方法整理或收集的数据集来对模型性能进行基准测试。作者选择的是以0.75的阈值来代表下游分析的二分类预测,它可以最好地划分positives和negatives的得分分布。
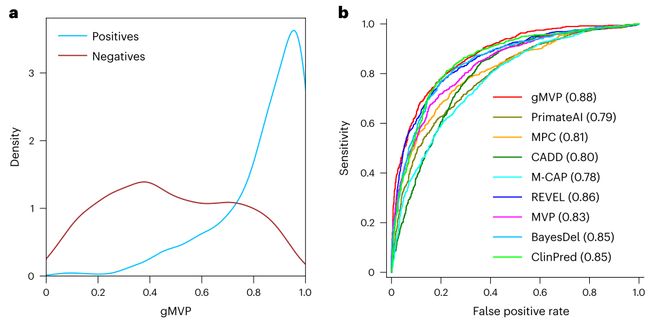
图2.gMVP与其他方法的测试性能评估
作者从数据库(HGMD,ClinVar and UniProt)中收集可能的致病性和良性错义突变分别作为positives和negatives训练,DiscovEHR中观察到的罕见错义突变作为额外的negatives训练。鉴于使用与训练数据同一来源随机划分的测试数据容易导致夸大性能,作者汇集了那些不太可能与训练数据共享相同系统错误的癌症体细胞突变用于测试。将其中为推断热点的错义突变作为positives,从DiscovEHR数据中随机选择的罕见突变作为negatives,这两种条件下gMVP得分的分布存在不同的模式(图2)。另外,与其他已发表的方法相比,gMVP的性能最好,其AUROC值为0.88,其次是REVEL为0.86。
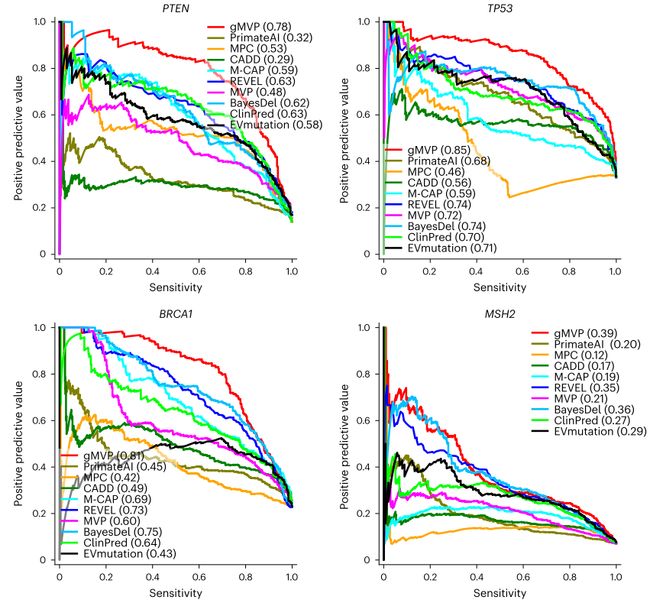
图3.gMVP和其他方法在识别已知疾病基因破坏性突变上的性能评估
作者还评估了gMVP和其他方法在预测同一基因中的破坏性突变和中性突变的能力。对4个已知的疾病风险基因TP53(540 positives,1108 negatives)、PTEN(262 positives,1632 negatives)、BRCA1(432 positives,1476 negatives)和MSH2(414 positives,5439 negatives)的深度突变进行扫描分析,所获得的功能性读出数据作为基本事实,这四个基因的所有变异数据都不在模型训练集之内。依据图3中precision-recall曲线图对比,结果证明了gMVP具有最好的预测效果。

图4.gMVP和其他方法在区分致病性与中性突变,以及预测离子通道基因中的GOF和LOF突变的性能评估
通过迁移学习,经过训练的gMVP模型可以针对遗传研究中更具体的任务进行进一步优化。作者尝试了通过迁移学习来对突变的作用模式进行具体分类(GOF和LOF)。从10个电压-门控钠离子通道和10个钙离子通道基因中获取1517个致病性突变和2328个中性突变。他们基于原始gMVP模型的权重重新训练了一个新的模型gMVP-TL1用于区分LOF/GOF(positives)和中性突变(negatives)。结果显示优于原来的gMVP和其他已发表的方法。作者同时也训练了gMVP-TL2,旨在具体区分LOF和GOF。结果显示gMVP-TL2也明显优于funNCion的性能(如图4)。这表明由迁移学习技术辅助的gMVP模型可以在非常有限的训练数据集下,准确地分类预测通道基因中的LOF和GOF。
总结
作者开发了gMVP——一种基于图注意神经网络的新方法来预测可导致功能性破坏的错义突变。它使用图注意神经网络来学习蛋白质序列和结构上下文的表示,能够在协同进化引导下有效地汇集三维空间中功能相关或潜在接近的远端氨基酸位置的预测信息。研究证明了gMVP方法在临床基因检测和新风险基因发现中具有实用性。最后,作者还针对模型分析总结了一些未来可以改进的方面:将人类群体中随机观察到的罕见突变数据加入模型训练;将Transformer组件和蛋白质三维结构整合到模型中;使用规模更大更多样化的人口基因组数据。
参考资料
Zhang, H., Xu, M.S., Fan, X. et al. Predicting functional effect of missense variants using graph attention neural networks. Nat Mach Intell 4, 1017–1028 (2022).
https://doi.org/10.1038/s42256-022-00561-w
代码
https://github.com/ShenLab/gMVP/
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


