scDrug:从scRNA-seq到药物反应预测
scRNA-seq技术允许在转录组水平上对数千个细胞进行测量。scRNA-seq正在成为研究肿瘤微环境中细胞成分及其相互作用的重要工具。scRNA-seq也被用于揭示肿瘤微环境模式与临床结果之间的关联,并在复杂组织中剖析药物治疗的细胞特异性效应。scRNA-seq的最新进展推动了疾病和治疗靶点生物标志物的发现。虽然已经提出了利用scRNA-seq数据的基因表达来预测药物反应的方法,但还需要一个从scRNA-seq分析到药物发现的集成工具。scDrug作为一个完整pipeline,包括生成scRNA-seq聚类和预测药物治疗方法。scDrug管道由三个主要模块组成:用于鉴定肿瘤细胞亚群的scRNA-seq分析、细胞亚群的功能注释和药物反应预测。
来自:scDrug: From single-cell RNA-seq to drug response prediction
项目:https://github.com/ailabstw/scDrug
目录
- 背景概述
- 数据和方法
-
- scRNA-seq数据预处理
- 自动分辨率聚类
- 差异基因分析,细胞注释,功能富集
- 生存分析
- 药物反应预测
-
- 药物反应训练数据
- GEP作为特征
- 预测模型的框架
- 模型训练和验证
- 肝细胞癌病例分析
背景概述
scRNA-seq被用于分析高分辨率细胞组成,从而发现肿瘤异质性,并为定制化生物学任务提供了前所未有的机会。恶性肿瘤细胞表达特征的细节为药物治疗提供了靶点依据。药物重利用是基于已批准或正在研究的药物,开发针对不同疾病的治疗策略。为了连接药物发现和scRNA-seq分析两个领域,开发了scDrug。
scDrug可以从scRNA-seq分析到药物反应预测。在scDrug中,首先构建了scRNA-seq分析管道,用于对scRNA-seq数据进行全面分析。实现了在Python环境下对肿瘤细胞进行亚群聚类。
接下来,整合了两种不同的方法来预测针对癌细胞亚群的药物治疗,使用包括LINCS, GDSC和PRISM在内的公共数据集来全面表征癌细胞系的分子特征。具体来说,一种方法预测药物对特定肿瘤簇的敏感性,另一种方法预测药物对肿瘤簇的综合作用。scDrug提供预测结果,供领域专家对所选药物进行评价。实验结果表明,scDrug可以成功捕获细胞对药物治疗的反应。总之,scDrug允许研究人员探索肿瘤细胞的异质性,并找到有效治疗的候选药物。
数据和方法
scRNA-seq数据预处理
第一步是scRNA-seq数据分析,包括Scanpy的数据预处理、MAGIC对输入进行插补、Harmony进行批次校正、Louvain进行聚类、Scanpy差异表达基因DEG鉴定、GSEAPY功能富集注释,scMatch细胞类型注释。
在数据预处理中,过滤掉表达少于200个基因的细胞和表达少于3个细胞的基因,将线粒体基因占比的细胞保持在30%以下。剩余的数据进行归一化到每细胞10000个总数,再进行自然对数变换,高变基因搜索,缩放到单位方差和零均值。一旦需要数据输入,scDrug还集成了MAGIC来输入缺失值。接下来,应用主成分分析(PCA),并根据需要使用Harmony消除批次效应。然后,计算前20个主成分的邻居图,并使用Louvain算法将细胞聚类成组。
自动分辨率聚类
为了确定聚类的分辨率,用户可以选择手动或自动分配。在自动模式下,对间隔为0.2的[0.4,1.4]区间内的分辨率值计算了chooseR中描述的基于次抽样的鲁棒性评分。对于给定的分辨率,使用定义为1.0的距离矩阵减去5次聚类的共聚类频率来计算平均轮廓分数,每次聚类在数据集的80%的随机子集上执行,不进行替换。将得分最高的分辨率作为最优聚类分辨率。
差异基因分析,细胞注释,功能富集
聚类后,scDrug使用默认参数的scanpy函数rank_genes_groups对每个聚类的基因进行排序,以识别DEGs。然后,scDrug用GSEAPY进行功能富集。此外,使用human GO_Biological_Process_2021库对log2倍变化大于2且p值和调整后的p值均低于0.01的DEG执行enrichment。对于细胞类型注释,使用所有基因表达,并计算其细胞的平均表达量作为每个簇的基因表达谱(GEP,gene expression profile)。接下来,应用scMatch,根据参考数据集的GEP对簇进行注释。
基于scRNA-seq数据分析的输出,包括一个AnnData对象,一个基因表达谱GEP,批校正、聚类和细胞类型注释结果的UMAPs,以及DEGs和GSEA文件。
生存分析
为了预测每个聚类对患者生存的影响,首先选择每个簇的前20个差异表达基因作为簇的特异性基因签名。然后,从TCGA数据库中下载不同癌症患者的bluk RNA profiles和相应的临床信息。为了评估每个患者的肿瘤簇活性,为每个患者构建了一个expression table,每个列代表一个簇的基因特征。对于每一簇及其所选的20个基因中的某一个,如果该患者的该基因表达高于所有患者的中位数表达,则赋值为1;否则,该值设置为0。按列求和(以下称为“activity score”)表示患者中每个簇的激活水平。对于每一簇,如果患者的活动得分在最高或最低四分位数,则将其分为“高表达”簇和“低表达”簇。最后,用Kaplan-Meier曲线和log-rank分析的p值比较两组的生存率(图S1)。下面示例中以A类型细胞为例,对比了A簇激活高低的两组患者,然后结合临床信息得到生存曲线。这里的生存分析是为了找到疾病相关的簇。

- 图S1:从 TCGA 中评估了 scRNA-seq 数据识别的每种细胞类型特异性特征的表达水平。 对于每个簇(细胞类型),将患者分为两组,一组为高表达,另一组为低表达。最后,用 Kaplan-Meier 曲线和 log rank p 值分析比较了这两组的生存情况。
药物反应预测
在scDrug管道中,使用第一步生成的AnnData对象,并应用CaDRReS-Sc(Predicting heterogeneity in clone-specific therapeutic vulnerabilities using single-cell transcriptomic signatures)进行药物反应预测。CaDRReS-Sc是一个基于scRNA-seq数据的强大的癌症药物反应预测的机器学习框架,它估计细胞簇的halfmaximal inhibitory concentration(IC50)。基于CaDRReSSc框架,提供了两种预训练的预测模型GDSC和PRISM,用于预测细胞簇的药物反应。
这两个模型是使用GDSC和PRISM数据集的基因表达和药物反应数据,通过无样本偏差的目标函数进行训练的。通过计算实际药物反应值和预测药物反应值的Spearman相关系数来评估预测性能。按照升序排列,scDrug剔除了drug-wise系数低于第一个四分位数系数的药物。
“IC50”(半数最大抑制浓度):表示在一定浓度下,化合物能够抑制蛋白质活性的程度
药物反应训练数据
对于GDSC模型,scDrug使用了226种药物在1074种癌细胞系中的反应数据(测量的IC50),数据来源于CaDRReS-Sc的GDSC数据集,GDSC数据集作为训练数据集。对于PRISM模型,scDrug使用PRISM Repurposing数据集(19Q4版本)作为训练数据,该数据集包含1448种药物对480种细胞系的反应。PRISM数据集以剂量-反应曲线下的面积(AUC)提供药物反应(不是来自IC50)。
GEP作为特征
对于GDSC模型,我们使用GDSC数据库中CaDRReS-Sc提供的1018个癌细胞系的基因表达数据,选择所有细胞系中共有的17419个基因作为特征基因进行模型训练。对于PRISM模型,从DepMap Portal(https://depmap.org/portal/)下载CCLE(Cancer Cell Line Encyclopedia)表达数据(21Q3版本),包含1,379个细胞系和19,177个基因。选择表达与PRISM AUC相关且绝对Pearson相关系数至少为0.2的8087个基因作为特征基因。scDrug计算了每个特征基因在细胞系间平均表达量的log2表达倍数变化。
预测模型的框架
为了预测细胞簇的IC50,scDrug计算了相对于AnnData的平均基因表达值的log2倍变化,并预测了每个细胞的IC50值。然后,平均IC50预测值确定每个簇的IC50。或者利用簇和其他簇之间的log2倍变化,直接预测簇的IC50。
模型从转录组学和药物反应中学习了潜在的药物-基因组学关系。CaDRReS-Sc中提出的模型定义为: s ^ i u = μ + b i Q + b u P + q i ⋅ p u = μ + b i Q + b u P + q i ( x u W P ) T \widehat{s}_{iu}=\mu+b_{i}^{Q}+b_{u}^{P}+q_{i}\cdot p_{u}=\mu+b_{i}^{Q}+b_{u}^{P}+q_{i}(x_u W_{P})^{T} s iu=μ+biQ+buP+qi⋅pu=μ+biQ+buP+qi(xuWP)T其中, s i u s_{iu} siu是药物 i i i对细胞系 u u u的观测药物反应(IC50), s ^ i u \widehat{s}_{iu} s iu表示预测的药物反应, μ \mu μ为总体平均药物反应, b i Q b_{i}^{Q} biQ和 b u P b_{u}^{P} buP分别是药物 i i i和细胞系 u u u的偏置项, q i , p u ∈ R f q_{i},p_{u}\in R^{f} qi,pu∈Rf表示药物 i i i和细胞系 u u u在latent space下的f-dim表征。 W P ∈ R d × f W_{P}\in R^{d\times f} WP∈Rd×f是将基因表达水平 x u ∈ R d x_u\in R^{d} xu∈Rd投影到latent space的变换矩阵, d d d为基因数。也有简化的: s ^ i u = b i Q + q i ⋅ p u \widehat{s}_{iu}=b_{i}^{Q}+q_{i}\cdot p_{u} s iu=biQ+qi⋅pu,目标函数定义为: L ( θ ) = 1 2 K [ ∑ i ∑ u ( s i u − s ^ i u ) 2 + λ ∑ d ∣ ∣ w d ∣ ∣ 2 + λ ∑ i ∣ ∣ q i ∣ ∣ 2 ] L(\theta)=\frac{1}{2K}[\sum_{i}\sum_{u}(s_{iu}-\widehat{s}_{iu})^{2}+\lambda\sum_{d}||w_{d}||^{2}+\lambda\sum_{i}||q_{i}||^{2}] L(θ)=2K1[i∑u∑(siu−s iu)2+λd∑∣∣wd∣∣2+λi∑∣∣qi∣∣2]其中, K K K是drug-cell pairs的总数, λ \lambda λ是L2正则化系数, w d w_d wd是 W P W_{P} WP中的向量。模型预测流程见图S2。

- 图S2:该图显示了训练 PRISM 药物反应模型并将其用于下游应用。对于训练任务,目标是最小化预测药物反应值和真实药物反应值(IC50)之间的损失。 对于应用任务,使用训练表达谱计算特征并用于预测药物反应。CCLE:Cancer Cell Line Encyclopedia。
模型训练和验证
PRISM和GDSC模型分别使用140维和10维潜在空间进行训练,学习率为0.01,最大epoch设置为100,000。为了评估unseen细胞系的性能,scDrug将24个细胞系作为验证集,并计算它们的中位数绝对误差,以及实际和预测药物反应之间的Pearson相关系数。
基于CaDRReS-Sc预测药物反应是一种候选药物预测方式,还有另一种是基于Premnas计算框架,结合LINCS L1000实现联合药物治疗的方案。两者分别有以下思想指导:
- 基于CaDRReS-Sc预测药物反应:学习药物与细胞系的IC50数据,所以在搜索候选药物时,是根据IC50来筛选的。
- Premnas:基于LINCS L1000学习扰动,扰动结果体现在施加药物扰动后,各个细胞系中细胞数量的变化,肿瘤细胞系的数量变少,说明该扰动是有利的。
总体而言,scDrug搜索药物是基于生存分析确定癌细胞系,基于药物和细胞系的统计数据来学习的,没有涉及到具体基因,蛋白层面,不能与现在的CPI模型相结合
肝细胞癌病例分析
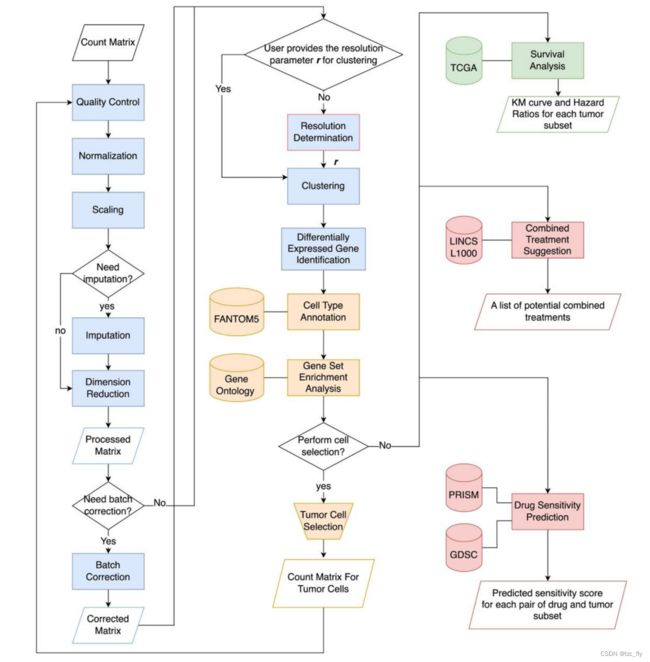
- 图3:scDrug工作流程。scDrug的第一部分分析scRNA-seq以生成细胞簇(蓝色)。scDrug的第二部分执行细胞类型和功能注释(黄色)。scDrug的第三部分运行生存分析,以帮助识别恶性肿瘤细胞簇(绿色),最后用两种不同的方法预测候选药物(红色)。
- 图4:肝细胞癌病例分析。
- A 肝细胞癌的scrna序列来自Sharma,2020;
- B Harmony批次校正后patientID分配的UMAP;
- C UMAP用于细胞聚类
- D scDrug自动计算分辨率;
- E 来自C的潜在肿瘤细胞亚聚类
- F 细胞簇gene ontology annotation的基因集富集分析
- G 用于生存分析的KM曲线示例
- H 热图显示PRISM数据库中通过CaDRReS-Sc预测抑制细胞生长的潜在药物。每个单元格代表肿瘤细胞簇对药物的预测敏感性评分值
- I 根据Premnas预测,LINCS L1000数据库中六种药物的最佳联合治疗方案的热图,以杀死最多数量的细胞簇。