分子动力学模拟-gromacs的基本使用
由pdb文件生成拓扑文件
本文档转写自gromacs教程,用以记录学习过程
gromacs推荐使用conda安装:conda install -c bioconda gromacs
-
下载pdb文件:从RCSB
其检索号为1aki,wget下载即可 -
去除水分子
对于不需要水分子的模拟,需要用命令除去水分子grep -v HOH 1aki.pdb > 1AKI_clean.pdb -
生成拓扑文件
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce #生成三个文件; 包含力场信息的拓扑文件 包含限制信息的文件 处理后的结构文件 -
力场的选择(共有15个立场,输入数字后按回车确认)
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals) -
得到三个结果文件
1AKI_processed.gro(gromacs支持的结构文件), topol.top(拓扑文件,包含使用的力场信息,后续的相关参数也要对应), and posre.itp(含有限制信息的文件).
设置box和solvent溶剂
设置盒子
对于盒子的设置,本例使用的是正方体,但是正十二面体的菱形可能是一个更好的选择(能够使用更少的水分子作为填充)。
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
#-c表示蛋白放在box中间
#-d 表示蛋白边缘距离盒子边缘最小的距离为1nm,这个量同样适用于其他的模拟
#-bt 设置盒子的类型
填充溶剂
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
#-cs spc216.gro设置了溶剂的模型标准,是gromacs的通用标准。同样适用于其他非水的溶剂系统
#输出新的结构文件,并同时修改拓扑文件。
对于水分子,拓扑文件中会显示加入了多少个水分子, SOL项
按照电荷数量添加离子
使用grompp来生成一个.trp的输入文件,再用genion来将溶剂分子替换成相应的离子
之前的top文件的最后显示有8个电子,所以将填入相应数量的离子
下载配置文件
http://www.mdtutorials.com/gmx/lysozyme/Files/ions.mdp
运行命令:
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
#ions.mdp是一个关于优化参数的文件,应该是可以复用的
#输出相应的离子tpr文件
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
#-pname NA -nname CL 指定了填入的正负离子的标准名称及NaCl
#-neutra设定为中和系统(即系统的总带电量为0?)
最后的top文件显示
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10636
CL 8
能量最小化
能量的最小化能够保证初始结构的稳定,其过程类似于之前添加离子的步骤:grompp命令用参数文件.mdp生成相应的二进制输入文件.tpr,再使用mdrun命令来进行能量最小化。
能量最小化的参数配置文件下载。
http://www.mdtutorials.com/gmx/lysozyme/Files/minim.mdp
gmx grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
再使用以下命令来执行能量最小化
gmx mdrun -v -deffnm em
#-v 为打印选项
#-deffnm确定相应的结果文件名
生成以下四个文件
em.log: ASCII-text log file of the EM process
em.edr: Binary energy file
em.trr: Binary full-precision trajectory
em.gro: Energy-minimized structure
并在最后打印:
Steepest Descents converged to Fmax < 1000 in 778 steps
Potential Energy = -5.8640175e+05(要保证为负数)
Maximum force = 9.9928015e+02 on atom 736(数值要小于1000)
Norm of force = 2.1921333e+01
最后可以通过
gmx energy -f em.edr -o potential.xvg
来进行能量收敛的绘图
平衡
之前的溶剂和溶质的优化是分割的,所以我们还要使得溶剂和溶质同时优化到我们想要的状态(温度,压力)。
第一阶段
第一阶段是对于温度的平衡,在NVT系统中进行(constant Number of particles, Volume, and Temperature)。
同样要使用相关的配置文件.mdp
http://www.mdtutorials.com/gmx/lysozyme/Files/nvt.mdp
同样使用类似上一步能量最小化的命令来进行
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
#这一步耗时相当久,吃满所有核
其中mdp的一些参数如下所示:
gen_vel = yes: Initiates velocity generation. Using different random seeds (gen_seed) gives different initial velocities, and thus multiple (different) simulations can be conducted from the same starting structure.
tcoupl = V-rescale: The velocity rescaling thermostat is an improvement upon the Berendsen weak coupling method, which did not reproduce a correct kinetic ensemble.
pcoupl = no: Pressure coupling is not applied
更加详细的可以在手册中找到
最后也同样可以使用
gmx energy -f nvt.edr -o temperature.xvg
进行收敛情况的绘图
第二阶段
第二阶段是对于压力的平衡,在NPT系统中进行( the Number of particles, Pressure, and Temperature are all constant)。
同时是使用一个mdp参数文件,但其中一些参数将会改变
continuation = yes: We are continuing the simulation from the NVT equilibration phase
gen_vel = no: Velocities are read from the trajectory (see below)
使用的命令如下:
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
#-t生成checkpoint文件
#-c生成最后用的结果文件
gmx mdrun -deffnm npt
使用一下命令可视化其过程:
gmx energy -f npt.edr -o pressure.xvg
#18
gmx energy -f npt.edr -o density.xvg
#24
进行MD
同样下载使用mdp参数文件:
http://www.mdtutorials.com/gmx/lysozyme/Files/md.mdp
相应的参数设置需要查询手册
执行命令:
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
#将估计可能的计算了PME值
执行模拟
gmx mdrun -deffnm md_0_1
#默认是使用所有线程?
#使用GPU加速
gmx mdrun -deffnm md_0_1 -nb gpu
结果分析
- 轨迹矫正
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
#Select 1 ("Protein") as the group to be centered and 0 ("System") for output
- RMSD计算
gmx rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg -tu ns
#计算模拟后的
#Choose 4 ("Backbone") for both the least-squares fit and the group for RMSD calculation
gmx rms -s em.tpr -f md_0_1_noPBC.xtc -o rmsd_xtal.xvg -tu ns
#计算原晶体的
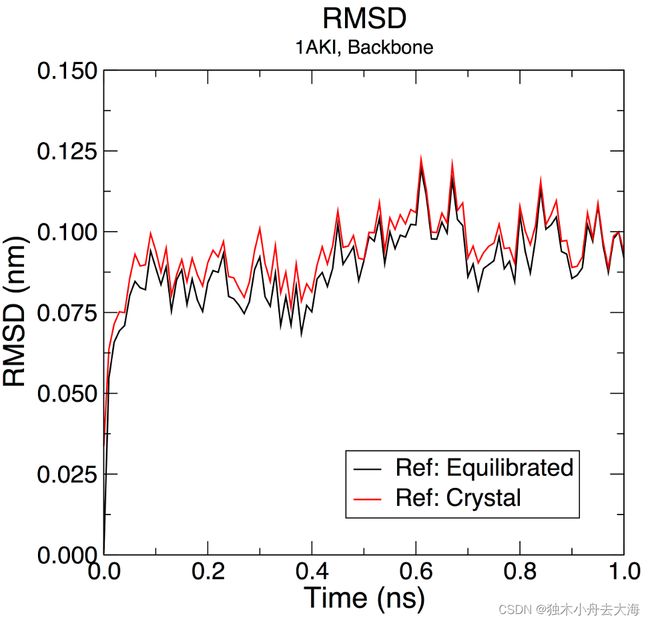
3. 计算蛋白的回转半径
回转半径可以用以衡量蛋白结构的紧实与否
gmx gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
#Choose group 1 (Protein) for analysis.
