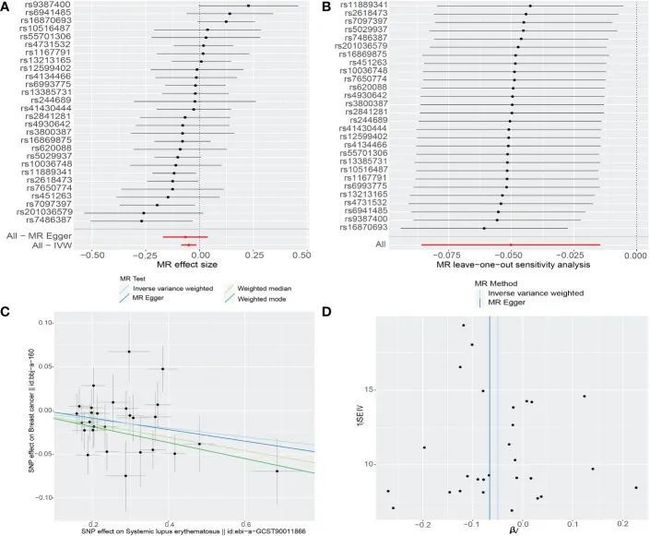
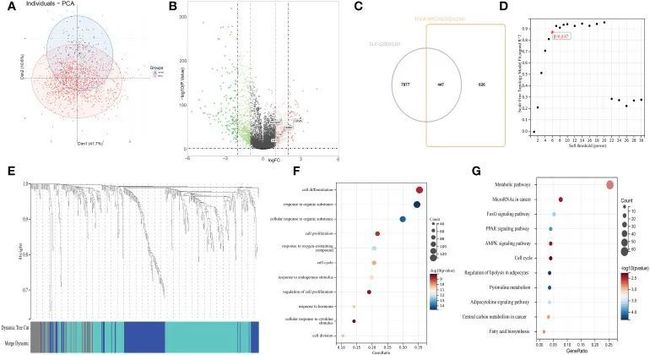
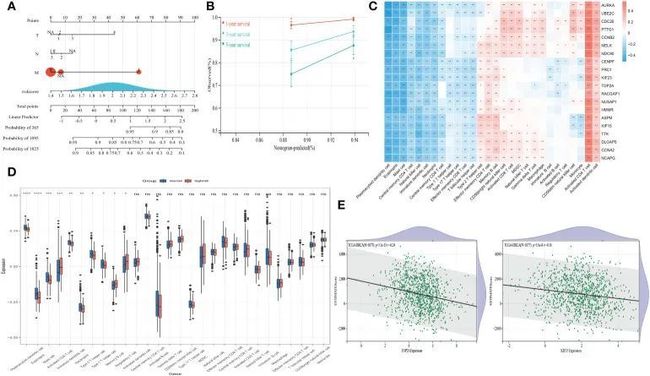
- 《互联网时代教师自主成长的模式研究》论文阅读与思考2
宁超群
2.第二部分教师自主成长的模式建构,实质上是对新网师底层逻辑的描述。你认为,新网师的培训模式与传统常见的培训模式有哪些区别?这些区别有什么意义或价值?读完第二部分后,你对新网师有哪些新的认识或理解?你认为新网师目前哪些方面做得好,哪些方面做得还不够?答:我认为新网师的培训模式与传统常见的培训模式有以下区别:(1)培训对象的参与动机不同。新网师学员的参与是自觉自愿、积极主动,而传统培训更多是被迫参与
- 【定位系列论文阅读】-Patch-NetVLAD: Multi-Scale Fusion of Locally-Global Descriptors for Place Recognition(一)
醉酒柴柴
论文阅读学习笔记
这里写目录标题概述研究内容Abstract第一段(介绍本文算法大致结构与优点)1.Introduction介绍第一段(介绍视觉位置识别的重要性)第二段(VPR的两种常见方法,本文方法结合了两种方法)第三段(本文贡献)第四段(为证明本文方法优越性,进行的测试以及比较)2.RelatedWork相关工作第一段(介绍早期与深度学习的全局图像描述符)第二段(介绍局部关键点描述符)第三段(局部描述符可以进一
- 论文阅读笔记(十九):YOLO9000: Better, Faster, Stronger
__Sunshine__
笔记YOLO9000detectionclassification
WeintroduceYOLO9000,astate-of-the-art,real-timeobjectdetectionsystemthatcandetectover9000objectcategories.FirstweproposevariousimprovementstotheYOLOdetectionmethod,bothnovelanddrawnfrompriorwork.Theim
- 论文阅读笔记: DINOv2: Learning Robust Visual Features without Supervision
小夏refresh
论文计算机视觉深度学习论文阅读笔记深度学习计算机视觉人工智能
DINOv2:LearningRobustVisualFeatureswithoutSupervision论文地址:https://arxiv.org/abs/2304.07193代码地址:https://github.com/facebookresearch/dinov2摘要大量数据上的预训练模型在NLP方面取得突破,为计算机视觉中的类似基础模型开辟了道路。这些模型可以通过生成通用视觉特征(即无
- 周四 2020-01-09 08:00 - 24:30 多云 02h10m
么得感情的日更机器
南昌。二〇二〇年一月九日基本科研[1]:1.论文阅读论文--二小时十分2.论文实现实验--小时3.数学SINS推导回顾--O分4.科研参考书【】1)的《》看0/0页-5.科研文档1)组织工作[1]:例会--英语能力[2]:1.听力--十分2.单词--五分3.口语--五分4.英语文档1)编程能力[2]:1.编程语言C语言--O分2.数据结构与算法C语言数据结构--O分3.编程参考书1)陈正冲的《C语
- 【论文阅读】Mamba:选择状态空间模型的线性时间序列建模(二)
syugyou
Mamba状态空间模型论文阅读
文章目录3.4一个简化的SSM结构3.5选择机制的性质3.5.1和门控机制的联系3.5.2选择机制的解释3.6额外的模型细节A讨论:选择机制C选择SSM的机制Mamba论文第一部分Mamba:选择状态空间模型的线性时间序列建模(一)3.4一个简化的SSM结构如同结构SSM,选择SSM是单独序列变换可以灵活地整合进神经网络。H3结构式最知名SSM结构地基础,其通常包括受线性注意力启发的和MLP交替地
- SAFEFL: MPC-friendly Framework for Private and Robust Federated Learning论文阅读笔记
慘綠青年627
论文阅读笔记深度学习
SAFEFL:MPC-friendlyFrameworkforPrivateandRobustFederatedLearning适用于私有和鲁棒联邦学习的MPC友好框架SAFEFL,这是一个利用安全多方计算(MPC)来评估联邦学习(FL)技术在防止隐私推断和中毒攻击方面的有效性和性能的框架。概述传统机器学习(ML):集中收集数据->隐私保护问题privacy-preservingML(PPML)采
- MixMAE(MixMIM):用于分层视觉变压器有效预训练的混合和掩码自编码器 论文阅读
皮卡丘ZPC
扩散模型阅读论文阅读
论文:MixMAE(arxiv.org)代码:Sense-X/MixMIM:MixMIM:MixedandMaskedImageModelingforEfficientVisualRepresentationLearning(github.com)摘要:本文提出MixMAE(MixedandmaskAutoEncoder),这是一种简单而有效的预训练方法,适用于各种层次视觉变压器。现有的分层视觉变
- 【论文阅读】LLM4CP: Adapting Large Language Models for Channel Prediction(2024)
Bosenya12
科研学习论文阅读语言模型人工智能信道预测时间序列
摘要Channelprediction(信道预测)isaneffectiveapproach(有效方法)forreducingthefeedback(减少反馈)orestimationoverhead(估计开销)inmassivemulti-inputmulti-output(大规模多输入输出)(m-MIMO)systems.However,existingchannelpredictionmet
- 【论文阅读】AugSteal: Advancing Model Steal With Data Augmentation in Active Learning Frameworks(2024)
Bosenya12
科研学习模型窃取论文阅读模型窃取模型提取数据增强主动学习
摘要Withtheproliferationof(随着)machinelearningmodels(机器学习模型)indiverseapplications,theissueofmodelsecurity(模型的安全问题)hasincreasinglybecomeafocalpoint(日益成为人们关注的焦点).Modelstealattacks(模型窃取攻击)cancausesignifican
- Bert系列:论文阅读Rethink Training of BERT Rerankers in Multi-Stage Retrieval Pipeline
凝眸伏笔
nlp论文阅读bertrerankerretrieval
一句话总结:提出LocalizedContrastiveEstimation(LCE),来优化检索排序。摘要预训练的深度语言模型(LM)在文本检索中表现出色。基于丰富的上下文匹配信息,深度LM微调重新排序器从候选集合中找出更为关联的内容。同时,深度lm也可以用来提高搜索索引,构建更好的召回。当前的reranker方法并不能完全探索到检索结果的效果。因此,本文提出了LocalizedContrast
- A Tutorial on Near-Field XL-MIMO Communications Towards 6G【论文阅读笔记】
Cc小跟班
【论文阅读】相关论文阅读笔记
此系列是本人阅读论文过程中的简单笔记,比较随意且具有严重的偏向性(偏向自己研究方向和感兴趣的),随缘分享,共同进步~论文主要内容:建立XL-MIMO模型,考虑NUSW信道和非平稳性;基于近场信道模型,分析性能(SNRscalinglaws,波束聚焦、速率、DoF)XL-MIMO设计问题:信道估计、波束码本、波束训练、DAMXL-MIMO信道特性变化:UPW➡NUSW空间平稳–>空间非平稳(可视区域
- 论文阅读:scMGCA----模型方法
dundunmm
论文阅读论文阅读人工智能聚类生物聚类单细胞聚类单细胞分析
Yu,Z.,Su,Y.,Lu,Y.etal.Topologicalidentificationandinterpretationforsingle-cellgeneregulationelucidationacrossmultipleplatformsusingscMGCA.NatCommun14,400(2023).https://doi.org/10.1038/s41467-023-36134
- 论文阅读:scHybridBERT
dundunmm
论文阅读机器学习人工智能神经网络深度学习单细胞基因测序
ZhangWei,WuChenjun,XingFeiyang,JiangMingfeng,ZhangYixuan,LiuQi,ShiZhuoxing,DaiQi,scHybridBERT:integratinggeneregulationandcellgraphforspatiotemporaldynamicsinsingle-cellclustering,BriefingsinBioinform
- 【论文阅读】Purloining Deep Learning Models Developed for an Ultrasound Scanner to a Competitor Machine
Bosenya12
科研学习模型窃取论文阅读深度学习人工智能模型安全
TheArtoftheSteal:PurloiningDeepLearningModelsDevelopedforanUltrasoundScannertoaCompetitorMachine(2024)摘要Atransferfunctionapproach(传递函数方法)hasrecentlyproveneffectiveforcalibratingdeeplearning(DL)algorit
- 《Motion Forecasting with Dual Consistency and Multi-Pseudo-Target Supervision》论文阅读之DCMS
山水之间2018
无人驾驶PaperReading大数据轨迹预测自动驾驶人工智能
目录摘要1简介2相关工作3.方法3.1结构3.2双重一致性约束3.3多伪目标监督3.4学习4实验4.1实验装置4.2实验结果4.3消融研究4.4泛化能力5限制6结论DCMS:具有双重一致性和多伪目标监督的运动预测香港科技大学暂无代码。摘要我们提出了一种具有双重一致性约束和多伪目标监督的运动预测新框架。运动预测任务通过结合过去的空间和时间信息来预测车辆的未来轨迹。DCMS的一个关键设计是提出双重一致
- 时序预测相关论文阅读笔记
能力越小责任越小YA
论文阅读笔记时序预测Transformer
笔记链接:【有道云笔记】读论文(记录)https://note.youdao.com/s/52ugLbot用于个人学习记录。
- 【论文阅读|cryoET】本周粗读汇总
吃吃今天努力学习了吗
冷冻电镜三维重建论文阅读
论文1:CryoDRGN-ET:深度重建生成网络以可视化细胞内动态生物分子Abstract虽然冷冻电子断层扫描可以以分子分辨率揭示结构,但图像处理算法仍然是解决原位生物分子结构异质性的瓶颈。本文介绍CryoDRGN-ET用于cryoET断层图的异质重建。CryoDRGN-ET直接从子断层扫描倾斜系列图像中学习三维密度图的深度生成模型,并且可以捕获成分和构象不同的状态。通过原位恢复肺炎支原体核糖体中
- Your Diffusion Model is Secretly a Zero-Shot Classifier论文阅读笔记
Rising_Flashlight
论文阅读笔记计算机视觉
YourDiffusionModelisSecretlyaZero-ShotClassifier论文阅读笔记这篇文章我感觉在智源大会上听到无数个大佬讨论,包括OpenAISora团队负责人,谢赛宁,好像还有杨植麟。虽然这个文章好像似乎被引量不是特别高,但是和AI甚至人类理解很本质的问题很相关,即是不是要通过生成来构建理解的问题,文章的做法也很巧妙,感觉是一些学者灵机一动的产物,好好学习一个!摘要这
- 【论文阅读】QUEEN: Query Unlearning against Model Extraction(2024)
Bosenya12
科研学习模型窃取论文阅读提取攻击模型安全
摘要Modelextractionattacks(模型提取攻击)currentlyposeanon-negligiblethreat(不可忽视的威胁)tothesecurity(安全性)andprivacy(隐私性)ofdeeplearningmodels.Byqueryingthemodelwithasmalldataset(通过小数据集查询模型)andusingthequeryresultsa
- 【论文阅读33】Deep learning optoacoustic tomography with sparse data
弹伦琴的雷登
【论文阅读系列】人工智能深度学习论文阅读图像处理
Deeplearningoptoacoustictomographywithsparsedata论文题目:基于稀疏数据的深度学习光声断层扫描论文链接:Deeplearningoptoacoustictomographywithsparsedata|NatureMachineIntelligence代码链接:GitHub-ndavoudi/sparse_artefact_unet数据链接:Data发
- 论文阅读瞎记(四) Cascade R-CNN: Delving into High Quality Object Detection 2017
码大哥
深度学习人工智能
概述在物体检测中1,IOU阈值被用于判定正负样本。在低IOU阈值比如0.5的状态下训练模型经常产生噪音预测,然而检测效果会随着IOU增加而降低。两个主要因素:1.训练时的过拟合,正样本指数消失2.检测器最优IOU与输入假设的不匹配。一个单阶段的物体检测器CascadeR-CNN被提出用于解决这些问题。网络由一个检测序列组成,这些序列训练时会伴随IOU增长从而对FP样本更加有选择性地判别。检测器一个
- 【论文阅读】LLM4SGG: Large Language Models for Weakly Supervised Scene Graph Generation
进击的乔洋
论文阅读语言模型人工智能计算机视觉
【论文阅读】LLM4SGG:LargeLanguageModelsforWeaklySupervisedSceneGraphGenerationabstract由于全监督方法严重依赖昂贵标注,最近弱监督场景图生成(WSSGG)研究替代方案出现。在这一点上(Inthisregard),针对WSSGG的研究主要利用图像标题(imagecaption)来获取非局部三元组,而主要关注将非局部三元组建立在图
- Code Llama: Open Foundation Models for Code论文阅读
yang_daxia
大模型llamacodellama
整体介绍CodeLlama发布了3款模型,包括基础模型、Python专有模型和指令跟随模型,参数量分别为7B、13B、34B和70B。这些模型在长达16ktokens的序列上训练。都是基于Llama2。作者针对infilling(FIM)、长上下文、指令专门做了微调long-contextfine-tuning(LCFT).codellama细节CodeLlama模型家族初始化:所有CodeLla
- 【论文阅读】Model Stealing Attacks Against Inductive Graph Neural Networks(2021)
Bosenya12
科研学习模型窃取论文阅读图神经网络模型窃取
摘要Manyreal-worlddata(真实世界的数据)comeintheformofgraphs(以图片的形式).Graphneuralnetworks(GNNs图神经网络),anewfamilyofmachinelearning(ML)models,havebeenproposedtofullyleveragegraphdata(充分利用图数据)tobuildpowerfulapplicat
- VIT论文阅读: A Image is Worth 16x16 Words
Undefined游侠
论文阅读
简介在2024年,大家都知道了transformer的故事,但是在4年前,CNN和Transformer谁才是CV的未来,还没有那么确定。在简介部分,作者提到了一个令人失望的事实,在基于imagenet的实验中发现,transformer的表现差于同尺寸的ResNet。作者把原因归结到biastranslationequivarianceandlocality,这些CNN具有,但是transfor
- 【论文阅读】GLiRA: Black-Box Membership Inference Attack via Knowledge Distillation
Bosenya12
模型窃取科研学习论文阅读知识蒸馏成员推理攻击黑盒
摘要While(虽然)DeepNeuralNetworks(DNNs)havedemonstratedremarkableperformanceintasksrelatedtoperception(感知)andcontrol(控制),therearestillseveralunresolvedconcerns(未解决的问题)regardingtheprivacyoftheirtrainingdat
- 【论文阅读】APMSA: Adversarial Perturbation Against Model Stealing Attacks(2023)
Bosenya12
科研学习模型窃取论文阅读模型窃取防御对抗性扰动
摘要TrainingaDeepLearning(DL)model(训练深度学习模型)requiresproprietarydata(专有数据)andcomputing-intensiveresources(计算密集型资源).Torecouptheirtrainingcosts(收回训练成本),amodelprovidercanmonetizeDLmodelsthroughMachineLearni
- Conditional Flow Matching: Simulation-Free Dynamic Optimal Transport论文阅读笔记
猪猪想上树
论文阅读笔记
ConditionalFlowMatching:Simulation-FreeDynamicOptimalTransport笔记发现问题连续正规化流(CNF)是一种有吸引力的生成式建模技术,但在基于模拟的最大似然训练中受到了限制。解决问题介绍一种新的条件流匹配(CFM),一种针对CNFs的免模拟训练目标。具有稳定的回归目标,用于扩散模型中的随机流,但享有确定性流模型的有效推断。与扩散模型和CNF目
- 《论文阅读》EmpDG:多分辨率交互式移情对话生成 COLING 2020
365JHWZGo
情感对话论文阅读共情回复回复生成对话系统多分辨率对抗学习
《论文阅读》EmpDG:多分辨率交互式移情对话生成COLING2020前言简介模型架构共情生成器交互鉴别器损失函数前言亲身阅读感受分享,细节画图解释,再也不用担心看不懂论文啦~无抄袭,无复制,纯手工敲击键盘~今天为大家带来的是《EmpDG:Multi-resolutionInteractiveEmpatheticDialogueGeneration》出版:COLING时间:2020类型:共情回复关
- ASM系列五 利用TreeApi 解析生成Class
lijingyao8206
ASM字节码动态生成ClassNodeTreeAPI
前面CoreApi的介绍部分基本涵盖了ASMCore包下面的主要API及功能,其中还有一部分关于MetaData的解析和生成就不再赘述。这篇开始介绍ASM另一部分主要的Api。TreeApi。这一部分源码是关联的asm-tree-5.0.4的版本。
在介绍前,先要知道一点, Tree工程的接口基本可以完
- 链表树——复合数据结构应用实例
bardo
数据结构树型结构表结构设计链表菜单排序
我们清楚:数据库设计中,表结构设计的好坏,直接影响程序的复杂度。所以,本文就无限级分类(目录)树与链表的复合在表设计中的应用进行探讨。当然,什么是树,什么是链表,这里不作介绍。有兴趣可以去看相关的教材。
需求简介:
经常遇到这样的需求,我们希望能将保存在数据库中的树结构能够按确定的顺序读出来。比如,多级菜单、组织结构、商品分类。更具体的,我们希望某个二级菜单在这一级别中就是第一个。虽然它是最后
- 为啥要用位运算代替取模呢
chenchao051
位运算哈希汇编
在hash中查找key的时候,经常会发现用&取代%,先看两段代码吧,
JDK6中的HashMap中的indexFor方法:
/**
* Returns index for hash code h.
*/
static int indexFor(int h, int length) {
- 最近的情况
麦田的设计者
生活感悟计划软考想
今天是2015年4月27号
整理一下最近的思绪以及要完成的任务
1、最近在驾校科目二练车,每周四天,练三周。其实做什么都要用心,追求合理的途径解决。为
- PHP去掉字符串中最后一个字符的方法
IT独行者
PHP字符串
今天在PHP项目开发中遇到一个需求,去掉字符串中的最后一个字符 原字符串1,2,3,4,5,6, 去掉最后一个字符",",最终结果为1,2,3,4,5,6 代码如下:
$str = "1,2,3,4,5,6,";
$newstr = substr($str,0,strlen($str)-1);
echo $newstr;
- hadoop在linux上单机安装过程
_wy_
linuxhadoop
1、安装JDK
jdk版本最好是1.6以上,可以使用执行命令java -version查看当前JAVA版本号,如果报命令不存在或版本比较低,则需要安装一个高版本的JDK,并在/etc/profile的文件末尾,根据本机JDK实际的安装位置加上以下几行:
export JAVA_HOME=/usr/java/jdk1.7.0_25
- JAVA进阶----分布式事务的一种简单处理方法
无量
多系统交互分布式事务
每个方法都是原子操作:
提供第三方服务的系统,要同时提供执行方法和对应的回滚方法
A系统调用B,C,D系统完成分布式事务
=========执行开始========
A.aa();
try {
B.bb();
} catch(Exception e) {
A.rollbackAa();
}
try {
C.cc();
} catch(Excep
- 安墨移动广 告:移动DSP厚积薄发 引领未来广 告业发展命脉
矮蛋蛋
hadoop互联网
“谁掌握了强大的DSP技术,谁将引领未来的广 告行业发展命脉。”2014年,移动广 告行业的热点非移动DSP莫属。各个圈子都在纷纷谈论,认为移动DSP是行业突破点,一时间许多移动广 告联盟风起云涌,竞相推出专属移动DSP产品。
到底什么是移动DSP呢?
DSP(Demand-SidePlatform),就是需求方平台,为解决广 告主投放的各种需求,真正实现人群定位的精准广
- myelipse设置
alafqq
IP
在一个项目的完整的生命周期中,其维护费用,往往是其开发费用的数倍。因此项目的可维护性、可复用性是衡量一个项目好坏的关键。而注释则是可维护性中必不可少的一环。
注释模板导入步骤
安装方法:
打开eclipse/myeclipse
选择 window-->Preferences-->JAVA-->Code-->Code
- java数组
百合不是茶
java数组
java数组的 声明 创建 初始化; java支持C语言
数组中的每个数都有唯一的一个下标
一维数组的定义 声明: int[] a = new int[3];声明数组中有三个数int[3]
int[] a 中有三个数,下标从0开始,可以同过for来遍历数组中的数
- javascript读取表单数据
bijian1013
JavaScript
利用javascript读取表单数据,可以利用以下三种方法获取:
1、通过表单ID属性:var a = document.getElementByIdx_x_x("id");
2、通过表单名称属性:var b = document.getElementsByName("name");
3、直接通过表单名字获取:var c = form.content.
- 探索JUnit4扩展:使用Theory
bijian1013
javaJUnitTheory
理论机制(Theory)
一.为什么要引用理论机制(Theory)
当今软件开发中,测试驱动开发(TDD — Test-driven development)越发流行。为什么 TDD 会如此流行呢?因为它确实拥有很多优点,它允许开发人员通过简单的例子来指定和表明他们代码的行为意图。
TDD 的优点:
&nb
- [Spring Data Mongo一]Spring Mongo Template操作MongoDB
bit1129
template
什么是Spring Data Mongo
Spring Data MongoDB项目对访问MongoDB的Java客户端API进行了封装,这种封装类似于Spring封装Hibernate和JDBC而提供的HibernateTemplate和JDBCTemplate,主要能力包括
1. 封装客户端跟MongoDB的链接管理
2. 文档-对象映射,通过注解:@Document(collectio
- 【Kafka八】Zookeeper上关于Kafka的配置信息
bit1129
zookeeper
问题:
1. Kafka的哪些信息记录在Zookeeper中 2. Consumer Group消费的每个Partition的Offset信息存放在什么位置
3. Topic的每个Partition存放在哪个Broker上的信息存放在哪里
4. Producer跟Zookeeper究竟有没有关系?没有关系!!!
//consumers、config、brokers、cont
- java OOM内存异常的四种类型及异常与解决方案
ronin47
java OOM 内存异常
OOM异常的四种类型:
一: StackOverflowError :通常因为递归函数引起(死递归,递归太深)。-Xss 128k 一般够用。
二: out Of memory: PermGen Space:通常是动态类大多,比如web 服务器自动更新部署时引起。-Xmx
- java-实现链表反转-递归和非递归实现
bylijinnan
java
20120422更新:
对链表中部分节点进行反转操作,这些节点相隔k个:
0->1->2->3->4->5->6->7->8->9
k=2
8->1->6->3->4->5->2->7->0->9
注意1 3 5 7 9 位置是不变的。
解法:
将链表拆成两部分:
a.0-&
- Netty源码学习-DelimiterBasedFrameDecoder
bylijinnan
javanetty
看DelimiterBasedFrameDecoder的API,有举例:
接收到的ChannelBuffer如下:
+--------------+
| ABC\nDEF\r\n |
+--------------+
经过DelimiterBasedFrameDecoder(Delimiters.lineDelimiter())之后,得到:
+-----+----
- linux的一些命令 -查看cc攻击-网口ip统计等
hotsunshine
linux
Linux判断CC攻击命令详解
2011年12月23日 ⁄ 安全 ⁄ 暂无评论
查看所有80端口的连接数
netstat -nat|grep -i '80'|wc -l
对连接的IP按连接数量进行排序
netstat -ntu | awk '{print $5}' | cut -d: -f1 | sort | uniq -c | sort -n
查看TCP连接状态
n
- Spring获取SessionFactory
ctrain
sessionFactory
String sql = "select sysdate from dual";
WebApplicationContext wac = ContextLoader.getCurrentWebApplicationContext();
String[] names = wac.getBeanDefinitionNames();
for(int i=0; i&
- Hive几种导出数据方式
daizj
hive数据导出
Hive几种导出数据方式
1.拷贝文件
如果数据文件恰好是用户需要的格式,那么只需要拷贝文件或文件夹就可以。
hadoop fs –cp source_path target_path
2.导出到本地文件系统
--不能使用insert into local directory来导出数据,会报错
--只能使用
- 编程之美
dcj3sjt126com
编程PHP重构
我个人的 PHP 编程经验中,递归调用常常与静态变量使用。静态变量的含义可以参考 PHP 手册。希望下面的代码,会更有利于对递归以及静态变量的理解
header("Content-type: text/plain");
function static_function () {
static $i = 0;
if ($i++ < 1
- Android保存用户名和密码
dcj3sjt126com
android
转自:http://www.2cto.com/kf/201401/272336.html
我们不管在开发一个项目或者使用别人的项目,都有用户登录功能,为了让用户的体验效果更好,我们通常会做一个功能,叫做保存用户,这样做的目地就是为了让用户下一次再使用该程序不会重新输入用户名和密码,这里我使用3种方式来存储用户名和密码
1、通过普通 的txt文本存储
2、通过properties属性文件进行存
- Oracle 复习笔记之同义词
eksliang
Oracle 同义词Oracle synonym
转载请出自出处:http://eksliang.iteye.com/blog/2098861
1.什么是同义词
同义词是现有模式对象的一个别名。
概念性的东西,什么是模式呢?创建一个用户,就相应的创建了 一个模式。模式是指数据库对象,是对用户所创建的数据对象的总称。模式对象包括表、视图、索引、同义词、序列、过
- Ajax案例
gongmeitao
Ajaxjsp
数据库采用Sql Server2005
项目名称为:Ajax_Demo
1.com.demo.conn包
package com.demo.conn;
import java.sql.Connection;import java.sql.DriverManager;import java.sql.SQLException;
//获取数据库连接的类public class DBConnec
- ASP.NET中Request.RawUrl、Request.Url的区别
hvt
.netWebC#asp.nethovertree
如果访问的地址是:http://h.keleyi.com/guestbook/addmessage.aspx?key=hovertree%3C&n=myslider#zonemenu那么Request.Url.ToString() 的值是:http://h.keleyi.com/guestbook/addmessage.aspx?key=hovertree<&
- SVG 教程 (七)SVG 实例,SVG 参考手册
天梯梦
svg
SVG 实例 在线实例
下面的例子是把SVG代码直接嵌入到HTML代码中。
谷歌Chrome,火狐,Internet Explorer9,和Safari都支持。
注意:下面的例子将不会在Opera运行,即使Opera支持SVG - 它也不支持SVG在HTML代码中直接使用。 SVG 实例
SVG基本形状
一个圆
矩形
不透明矩形
一个矩形不透明2
一个带圆角矩
- 事务管理
luyulong
javaspring编程事务
事物管理
spring事物的好处
为不同的事物API提供了一致的编程模型
支持声明式事务管理
提供比大多数事务API更简单更易于使用的编程式事务管理API
整合spring的各种数据访问抽象
TransactionDefinition
定义了事务策略
int getIsolationLevel()得到当前事务的隔离级别
READ_COMMITTED
- 基础数据结构和算法十一:Red-black binary search tree
sunwinner
AlgorithmRed-black
The insertion algorithm for 2-3 trees just described is not difficult to understand; now, we will see that it is also not difficult to implement. We will consider a simple representation known
- centos同步时间
stunizhengjia
linux集群同步时间
做了集群,时间的同步就显得非常必要了。 以下是查到的如何做时间同步。 在CentOS 5不再区分客户端和服务器,只要配置了NTP,它就会提供NTP服务。 1)确认已经ntp程序包: # yum install ntp 2)配置时间源(默认就行,不需要修改) # vi /etc/ntp.conf server pool.ntp.o
- ITeye 9月技术图书有奖试读获奖名单公布
ITeye管理员
ITeye
ITeye携手博文视点举办的9月技术图书有奖试读活动已圆满结束,非常感谢广大用户对本次活动的关注与参与。 9月试读活动回顾:http://webmaster.iteye.com/blog/2118112本次技术图书试读活动的优秀奖获奖名单及相应作品如下(优秀文章有很多,但名额有限,没获奖并不代表不优秀):
《NFC:Arduino、Andro