铁死亡调控机制新发现——癌症篇
随着癌细胞对凋亡产生抗性,非细胞凋亡死亡模式的铁死亡成为对抗治疗耐药癌症的新策略,对传统疗法产生耐药性的细胞或转移性癌细胞已被证明对铁死亡的敏感性增加。因此,靶向癌症中的铁死亡调控元件可能提供新的治疗机会。
今年5月来自德国维尔茨堡大学的José Pedro Friedmann Angeli等人在《Trends in Cell Biology》上联合发表了题为“Ferroptosis: mechanisms and implications for cancer development and therapy response”的综述。该文对已知的铁死亡调控网络进行了概述,探讨它们在癌症的相关研究中的作用。通过对新兴的铁死亡调控途径的理解,为创新的抗癌策略的发展提供了希望。

01 克服抗癌抵抗的细胞死亡途径
癌细胞已经进化出多种策略来逃避内在的凋亡诱导机制,并利用凋亡抑制机制对标准治疗产生耐药性,导致耐药细胞的出现。目前,细胞死亡的非凋亡途径——铁死亡的研究,在根除抗凋亡癌细胞的研究中大有前景。
铁死亡是一种受调控的细胞死亡过程,其特征是铁催化的脂质过氧化产物的过量积累。铁死亡存在不同的调节机制来维持稳态,不受控制的铁死亡与各种病理条件有关,包括癌症、神经退行性变、中风、肾脏和视网膜损伤以及感染。"铁死亡"这一词最初是由Dixon及其同事在2012年创造的,用来描述由erastin引发的一种独特的细胞死亡形式,erastin是一种小分子化合物,能诱导表达大鼠肉瘤病毒(Ras)致癌基因的同源癌细胞系的细胞死亡。此后,将铁死亡和肿瘤发生联系起来的证据越来越多,特别是由于去分化和持久性癌细胞状态对铁死亡的脆弱性增加。

铁死亡调控要素
02 铁:一种不寻常的导致细胞死亡的催化剂
活性氧和氮源性循环(ROS)的过量产生会对包括膜脂在内的生物分子造成重大损害。
烷氧基或羟基自由基可以与含多不饱和脂肪酸(PUFA)的磷脂(PLs)通过Fenton反应形成脂质氢过氧化物和其他携带自由基的脂质。该反应由亚铁(Fe2+)催化,将过氧化氢(H2O2)转化为羟基自由基,触发脂质过氧化。此外,脂质氢过氧化物可以作为类Fenton反应的底物,产生烷氧基,引发脂质过氧化。
因此,铁利用率和稳态的调节是刺激脂质过氧化的关键成分,游离铁在Fenton反应中活性中间体的产生发挥着重要作用。最近的研究强调了铁代谢在肿瘤发生中的基本作用,以及靶向调节铁稳态的元素触发铁死亡和消除癌细胞的潜力。
铁在细胞外 Fe3+ 离子的形式进入细胞,与转铁蛋白结合。转铁蛋白受体将复合物内吞,铁被输送到溶酶体,并以还原的活性亚铁(Fe2+)状态暂时储存。亚铁从溶酶体中输出,提供了一个细胞内不稳定的LIP。在细胞体中,LIP可以被运送到细胞器中,通过铁蛋白转运到细胞外,或储存在铁蛋白笼中以限制其反应活性。自噬介导的铁蛋白降解使铁蛋白结合的铁再次可用。铁一旦可用便可以调节肿瘤发展过程中的几个关键过程,如与肿瘤微环境的相互作用和驱动癌症进展的表观遗传学修饰。
此外,铁过载与治疗压力下获得的癌症代谢可塑性有关,为选择难治性的细胞提供了克隆优势。相比之下,使用铁霉素(ironomycin)诱导溶酶体保留铁,可诱导乳腺CSCs的溶酶体膜通透和铁死亡,表明其对铁的依赖性。这些研究说明了铁代谢在特定癌症状态下的关键作用和针对铁代谢的可行性,以及利用其引发脂质过氧化以获得治疗效果的潜力。
03 铁死亡:通过细胞膜酸败杀死细胞
虽然脂质过氧化是生物系统中氧化失衡的一个明确来源,但触发膜酸败的确切机制仍不清楚。已观察到脂氧合酶(LOX)酶通过增加脂质氢过氧化物的细胞池来启动铁死亡,但它们的作用仍然存在争议,因为许多癌细胞铁死亡的发生独立于LOX表达。最近的研究表明,内质网氧化还原酶有助于铁死亡的发生。这些氧化还原酶可能通过将电子从NADPH/NADH提供给氧气来触发铁死亡,从而产生H2O2。此外,已有研究表明脂质自氧化是一种自发的过氧自由基介导的链式反应,通过传播脂质过氧化,在驱动细胞死亡过程中发挥了关键作用。
最终,PLs的过氧化衍生物改变了膜的结构和性质,导致质膜的通透性质变和铁死亡。铁死亡可以通过PL过氧化产物的特征性积累与其他细胞死亡方式区分开来;它的功能可以通过铁螯合剂、亲脂性抗氧化剂或PUFAs的消耗受到抑制。
除了防止铁死亡的主要酶GPX4外,铁死亡还受几个后备系统的调节,如甲戊酸钠(MVA)途径、FSP1泛醌、四氢生物蝶呤(BH4)、二氢叶酸还原酶(DHFR)、反硫化途径。
04 半胱氨酸-GSH-GPX4:铁死亡的第一道防线
细胞已经进化出一种特殊的机制来防止和修复PLs的氧化损伤,因为它们对这种损伤具有固有敏感性。靶向这些途径是触发铁死亡的核心。例如,erastin可以通过抑制半胱氨酸-谷氨酸反向转运蛋白-Xc-系统,来诱导铁死亡。
半胱氨酸的耗竭会导致含硫醇抗氧化剂迅速流失,如谷胱甘肽(GSH)。GSH作为谷胱甘肽过氧化物酶4(GPX4)的辅助因子发挥其对铁死亡的保护作用,GPX4利用GSH来减少过氧化的PLs。使用Ras-selective-lethal-3(RSL3)直接抑制GPX4也已经被证明可以触发铁死亡,而不受GSH耗竭或系统Xc-活性的影响。RSL3共价修饰了GPX4活性部位的硒半胱氨酸(Sec),阻断了细胞中过氧化的PLs的修复系统。此外,当GSH匮乏时,GPX4在半胱氨酸周围表现出一个异生位点,RSL3也能在此结合,不仅抑制GPX4,而且还导致GPX4被降解。半胱氨酸饥饿、GSH耗竭和相关的脂质过氧化可以追溯到一个特定的GSH依赖性酶,建立了第一个也是最重要的铁死亡级联的调节轴。
05 硒、硒半胱氨酸和GPX4
硒蛋白,如GPX4,含有Sec,这是一种与半胱氨酸结构相似的氨基酸,但用一个硒(Se)原子取代了硫,这种替换使硒蛋白具有独特的生物化学和物理特性。将硒并入硒蛋白是一个复杂的、高能耗的过程。这个复杂的过程需要合成一个Sec特异性转移RNA(Sec-tRNA),随后在UGA密码子上重新编码并加入Sec ,Se主要以硒蛋氨酸(SeMet)的有机形式、Sec或无机形式(如硒酸盐/SeO4-2和亚硒酸盐/SeO3-2离子)在饮食中获得。
无论Se的形式如何,其代谢主要发生在肝脏,在肝脏中Se载体SELENOP的生物合成。SELENOP是一种具有多个Sec残基的蛋白质,可作为向肝外组织的传递系统。然后SELENOP被分泌到血液循环中,并被细胞表面受体识别,包括LRP8(也称为APOER2)和LRP2(也称为Megalin),用于受体介导的内吞。接下来,Se原子需要从Sec中去除,以允许Sec-tRNA的生物合成,并最终翻译新的硒蛋白。
硒蛋白的表达受到Se和Sec可用性的不同影响。分析表明,非必需的硒蛋白在饮食中缺硒时被下调,以便为更多的必需蛋白保留可用的硒,如GPX4。尽管这一层次结构被认为是静态的,但这一概念似乎过于简单,在癌细胞中可以被改变。最近的工作表明,敲除LRP8,一个Se缺乏的模型,会导致GPX4的严重损失,而不会严重影响其他硒蛋白。这强调了破坏Se代谢和硒蛋白翻译的潜在策略,使癌细胞对铁死亡敏感。
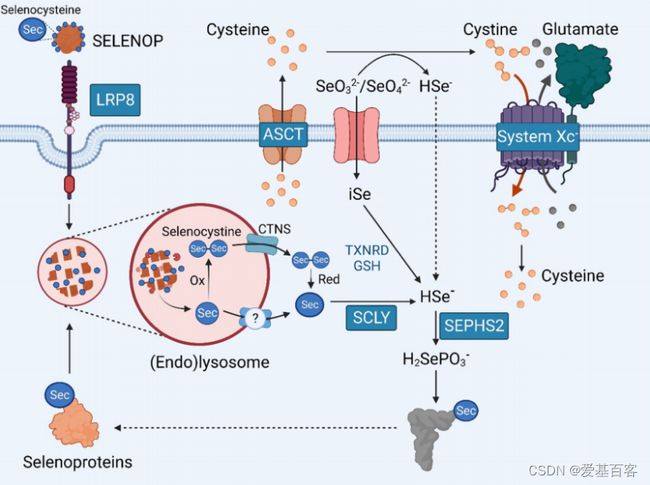
硒(Se)/硒半胱氨酸(Sec)的摄取和回收示意图
06 GPX4调控通路的新发现——文章解析
除了上述提到的GSH作为谷胱甘肽过氧化物酶4(GPX4)的辅助因子发挥其对铁死亡的保护作用,最新研究发现KLF11的过表达会抑制GPX4的转录。今年的5月份复旦大学中山医院胸外科团队在《Nature Communications Biology》上发表了名为“KLF11 regulates lung adenocarcinoma ferroptosis and chemosensitivity by suppressing GPX4”的文章,该文表示通过KLF11(一种转录因子)过表达可显著抑制肺癌发展,提高化疗效果。

肺腺癌(LUAD)是肺癌最常见的亚型,是世界范围内导致癌症死亡的主要原因。LUAD具有相对较强的增殖能力,生存率较差,化疗仍是这类恶性肿瘤的主要治疗方法之一。然而,对化疗的耐药性严重影响了LUAD患者的预后。因此,探索抑制LUAD增殖、提高LUAD化疗敏感性的方法势在必行。
该研究发现 KLF11 通过参与 GPX4 相关的铁死亡途径抑制肺腺癌细胞增殖并提高化疗敏感性。通过对用铁死亡诱导剂(FINs)预处理的 LUAD 细胞进行RNA-seq,发现在 FINs 处理的细胞中 KLF11 表达显着升高,表明 KLF11 可能参与铁死亡,通过蛋白水平的检测也证实了该推论。接下来,研究人员发现KLF11在LUAD细胞系中的表达明显低于在正常支气管上皮细胞中的表达。同时,KLF11还可增强化疗药物(CDDP和Pem)对LUAD细胞的毒性作用。由此表明KLF11促进了FINs诱导的铁死亡,抑制了细胞增殖,并增强了化疗药物在LUAD中的作用。

为了探索KLF11促进铁死亡的下游信号分子,作者分别对KLF11-NC和KLF11-OE细胞进行RNA-seq筛选出差异表达基因。然后,将RNA-seq、ChIP-Seq数据和特定铁死亡相关基因(从FerrDb数据库获得)取交集。结果表明,KLF11过表达后GPX4显著下调,推测GPX4可能是KLF11的靶标分子。通过qRT-PCR和western blot进一步验证了KLF11对GPX4表达的影响,发现过表达KLF11降低了GPX4水平,而敲除KLF11导致GPX4表达上调。其他铁死亡的关键调控因子,包括ACSL4和SLC7A11的水平没有受到影响。这些结果表明,KLF11可能通过调控GPX4的表达参与了LUAD的铁死亡。
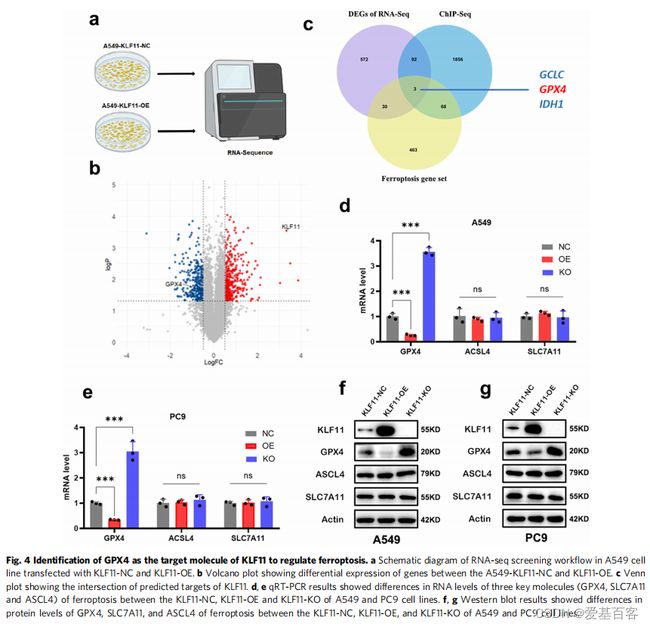
KLF11作为一种转录因子,作者推测其通过与启动子结合来转录抑制GPX4。通过ChIP-seq结果,发现在靠近转录起始位点(TSS)的GPX4启动子区域有一个非常强的klf11结合峰。ChIP-qPCR检测也显示,在A549和PC9细胞系(肺癌细胞)中,KLF11的结合均显著富集,由此证实了GPX4是KLF11的直接转录靶点。为了研究KLF11与GPX4启动子的结合是否具有功能,将人GPX4启动子序列克隆到pgl4-荧光素酶报告基因载体中,用于荧光素酶报告基因活性实验。结果揭示了KLF11与GPX4相互作用的机制,说明KLF11与GPX4启动子强结合,抑制GPX4转录。

随后通过小鼠疾病模型的构建,定期观察小鼠体内肿瘤的生长,发现恢复的 GPX4 表达可拮抗 KLF11 促进铁死亡、增加化疗敏感性和抑制体外和体内细胞增殖的能力。临床上数据分析显示,KLF11 在 LUAD 中下降,其低表达与患者生存率降低相关。该文的研究结果证实了 KLF11 促进 LUAD 中铁死亡的功能,从而抑制细胞增殖并增强化疗效果。
技术路线

上述关于铁死亡调控机制研究使用的ChIP-seq、ChIP-qPCR、RNA-seq等相关技术,爱基百客均可提供,且经验丰富。可提供一站式服务:包括前期方案设计、样本处理、测序、数据分析以及后期验证。欢迎各位老师咨询~
引用文献
-
Dos Santos AF,Fazeli G,Xavier da Silva TN, et al. Ferroptosis: mechanisms and implications for cancer development and therapy response. Trends Cell Biol. 2023;33 (12):1062-1076. doi:10.1016/j.tcb.2023.04.005
-
Zhao G,Liang J,Shan G, et al. KLF11 regulates lung adenocarcinoma ferroptosis and chemosensitivity by suppressing GPX4. Commun Biol. 2023;6 (1):570. doi:10.1038/s42003-023-04959-z