Seq-SetNet:直接利用多序列比对预测蛋白质二级结构
Seq-SetNet: directly exploiting multiple sequence alignment for protein secondary structure prediction
- Seq-SetNet:直接利用多序列比对预测蛋白质二级结构
-
- 1. 背景
-
- 1.1 当前问题
- 1.2 解决办法
- 2. 方法
-
- 2.1 多序列比对的信息表示
- 2.2 Seq-SetNet的体系结构
-
- 2.2.1 MSA编码器
- 2.2.2 聚合模块
- 2.2.3结构性能预测
- 2.3 模型训练过程
-
- 2.3.1 输入和输出
- 2.3.2 损失函数
- 3.实验结果
-
- 3.1 数据集和评价标准
- 3.2 蛋白质二级结构和扭转角的预测精度
- 3.3 Seq-SetNet模型各组成部分的贡献分析
-
- 3.3.1 Max函数的影响
- 3.3.2 MSA编码器的作用
- 3.3.3 多序列比对的影响
- 3.3.4 聚合模块中MAX函数的探索能力分析
- 3.3.5 与现有方法的比较
- 4. 总结
- 5. 补充
alignment for protein secondary structure prediction)
Seq-SetNet:直接利用多序列比对预测蛋白质二级结构
蛋白质结构的准确预测在很大程度上依赖于利用多序列比对(MSA)的残基突变和相关性,因为这个信息指定蛋白质的三级结构。目前广泛使用的预测方法通常将MSA转化为中间模型,如位置特定的得分矩阵或轮廓隐马尔可夫模型。然而,这些中间模型不能完全代表MSA携带的残基突变和相关性;因此,非常需要一种直接利用MSA有效方法。
我们报告了一种新的序列集网络(称为Seq-SetNet)直接和有效地利用MSA的蛋白质结构预测。
Seq-SetNet使用由两个关键元素组成的“编码和聚合”策略:
(i)编码模块,其将MSA中的组分(混合物中的各个成分)同源物作为输入,并将残基突变和相关性编码为每个残基的上下文特异性特征;
(ii)聚集模块,用于聚集从所有组分同源物中提取的特征,所述特征被进一步转化为查询蛋白质的残基的结构性质。
由于Seq-SetNet单独编码每个同源蛋白,因此它可以考虑插入和缺失以及残基之间的长距离相关性,从而比中间模型代表更多的信息。编码模块自动学习有效特征,从而避免了手动特征提取。Seq-SetNet使用对称聚集函数将同源蛋白作为序列集处理,使其预测结果与这些蛋白质的顺序一致。在流行的基准集上,我们展示了Seq-SetNet在预测残基的二级结构和扭转角方面的成功应用,提高了准确性和效率。
1. 背景
蛋白质三级结构预测的成功与否很大程度上依赖于查询蛋白质多重序列比对(MSA)的构建和利用。利用统计模型或深度神经网络构建高质量MSA已取得重大进展。对于查询蛋白质,其构建的MSA由其多个同源物组成,其残基排列在适当的位置以显示这些同源物与查询蛋白之间的突变。MSA中的同源蛋白质携带丰富的残基突变和残基-残基相关性信息,这些信息指定了查询蛋白质的三级结构。折叠蛋白质结构中的每个残基都有自己的结构环境,包括掩埋区、二级结构以及与其他残基的相互作用。这种结构环境,也称为结构背景,通常排除不相容的氨基酸类型,因此对相容的氨基酸类型显示出相当大的优先权。对相容氨基酸类型的优先形成MSA中同源蛋白质序列的特定残基分布。此外,两个或两个以上的残留接触通常会导致相应的MSA列之间显著相关。相反,残基突变的优先特征和相关性可以用来推断残基的结构环境。
1.1 当前问题
当前广泛的方法是将MSA转化为中间模型,比如PSSM(位置特征得分矩阵)或隐马尔可夫模型。但是中间模型无法完全代表MSA:
- PSSM会计算每个MSA列上的各种残基类型的出现,但并不能考虑残基之间的相关性;
- 隐马尔科夫模型可以描述残基之间的相关性,但其不容易按比例放大以揭示残基之间的远距离相关性;
因此,迫切需要一种有效的方法来直接、全面地从MSAs中提取与结构相关的信息。
神经网络在信息提取领域做得很好,尤其是图像处理领域。但是现有的神经网络框架并不适合MSA的特点,序列与图像的区别:
- MSA的分量序列本质上是一个无序的集合,因为任意两个序列的交换并不改变MSA所携带的信息。;相比之下,图像中的像素形成矩阵而不是集合; 因此,两行像素的交换通常会极大地改变图像;
- 此外,MSAS的组成蛋白质的数量和这些蛋白质的长度是高度可变的,而图像中的像素行总是具有相同的长度。;
这些独特的特性使得专门设计一个适合MSAS的框架变得非常可取。
1.2 解决办法
我们提出了这样一个神经网络框架(称为Seq-SetNet),直接利用MSA进行蛋白质结构预测。
Seq-SetNet使用由两个关键元素组成的“编码和聚合”策略:
(i)编码模块,其将MSA中的组分(混合物中的各个成分)同源物作为输入,并将残基突变和相关性编码为每个残基的上下文特异性特征;
(ii)聚集模块,用于整合从所有同源物中提取的特征,所述特征被进一步转化为查询蛋白质的残基的结构性质。
通过将编码模块单独应用于每个同源蛋白,Seq-SetNet具有比中间模型考虑更多信息的潜力。编码模块自动学习有效特征,从而避免了手动特征提取。聚集模块使用对称聚集函数将同源蛋白作为序列集处理,使其预测结果与这些蛋白质的顺序一致。
2. 方法
下图是Seq-SetNet的结构图。Seq-SetNet将查询蛋白的MSA作为输入,并报告查询蛋白的每个残基的结构特性作为预测结果。Seq-SetNet使用“编码和聚合”策略从MSA中提取残基突变和相关信息,然后基于这些信息推断残基的结构特性。这里,特征向量与残差的预测结构性质一起被示出为颜色条,其中具有大值的特征或结构性质以深色绘制,而具有小值的特征或结构性质以浅色绘制
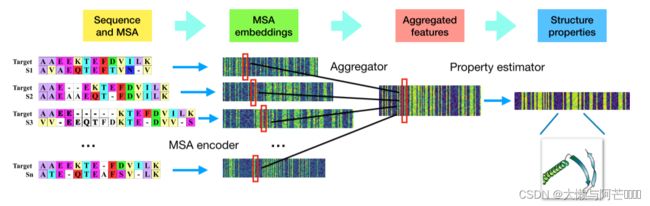
2.1 多序列比对的信息表示
对照结构图:为了减少信息损失,我们将MSA表示为一组比对,其中每个比对包含查询蛋白和同源蛋白。值得注意的是,在每个比对中,都添加了空位来代表缺失和插入。(虽然比对可能有不同的长度,我们的神经网络框架是专门设计来适应这样的比对)
我们将每个位置编码为44个元素的二元向量,其中包括查询蛋白质中残基的22个元素和同源蛋白质中比对残基的22个元素。这22个元素代表以one-hot向量形式表示,代表20种氨基酸、空位、未知氨基酸。
2.2 Seq-SetNet的体系结构
2.2.1 MSA编码器
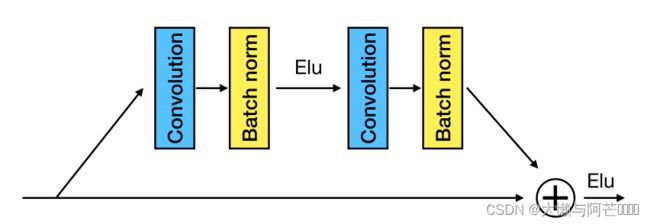
编码器相同且独立地处理每个单独的同源序列。对于给定MSA中的每个同源物,将相应的信息表示馈送到编码器中,从而产生每个位置的上下文特异性突变特征。为了使每个位置处的向量表示对其周围环境的信息进行编码,我们将编码器构造为具有8个残余块的一维卷积残差网络。每个残差块由与两个BatchNorm层交织的神经网络层组成。每个残差块由两个BatchNorm层、两个带64个滤波器的一维卷积层(核大小为3)和指数线性单元(ELU)非线性组成。
2.2.2 聚合模块
聚合模块旨在从MSA编码器提取的变异特征中聚合进化信息,在给出聚合模块的设计之前,我们首先描述了标记。 考虑具有L个残基的查询蛋白,和含有K个同源蛋白的预构建MSA。通过将编码模块应用于MSA中的第k个同源蛋白,我们已经获得了Lk×C 特征,表示为![]() ,其中Lk表示第k个序列对的长度,C表示编码模块的输出通道(本研究中为C=64 )。聚合模块的目的是提取这些嵌入,以获得进化特征,其具有的大小线性查询蛋白质的长度。对于查询蛋白质中的每个残基,我们期望获得固定大小的特征,这些特征可以充分描述该残基的进化情况。
,其中Lk表示第k个序列对的长度,C表示编码模块的输出通道(本研究中为C=64 )。聚合模块的目的是提取这些嵌入,以获得进化特征,其具有的大小线性查询蛋白质的长度。对于查询蛋白质中的每个残基,我们期望获得固定大小的特征,这些特征可以充分描述该残基的进化情况。
- 一个主要的困难是,嵌入的组件同源物通常有很大的不同,在他们的长度。为了解决这个问题,我们直接丢弃每个同源物的插入区域处的特征,即仅考虑与查询蛋白比对的位置。通过将此操作应用于第k个同系物的嵌入,我们得到了Lk×C 特征,表示为
 。 这样,所有嵌入都具有相同的大小。虽然我们丢弃了插入区域的特征,但是编码模块中的卷积层已经学习到相应的变异信息,并将其包含在对齐区域的特征中。
。 这样,所有嵌入都具有相同的大小。虽然我们丢弃了插入区域的特征,但是编码模块中的卷积层已经学习到相应的变异信息,并将其包含在对齐区域的特征中。 - 另一个重要的过程是在所有池嵌入中聚合有用的特性。 实际上,同源蛋白总是表现为一个集合,因此理想的聚合模块应该对MSA中同源蛋白的顺序免疫。为了满足这一原则,我们提出使用对称聚合函数,从所有同源蛋白的提取特征中取元素最大值。我们将聚合的特征表示为A∈R^Lk ×C^,并使用以下聚合函数计算它们:

其中A(i,j)表示查询蛋白质中第i个残基的第j个聚合特征,并且表示第k个同源物在位置i处的第j个提取的特征。注意,最大聚合函数是对称的;因此,聚合特征A不随提取特征x’1 ,x’2 ,x’3 ,…,x’k 的顺序而改变。 - Max函数在Seq-SetNet中发挥了重要作用。由于良好的数学性质,max函数被广泛应用于神经网络的设计中,如ReLU激活函数和max池化,它在许多计算机视觉任务上表现出了强大的能力。我们构建了一个可能的工作方式,以更好地理解为什么这样的架构可以完美地聚合所有的突变信息。在最坏的情况下当C足够大时,MSA编码器的工作方式类似于将不同的上下文特定的突变情况映射到不同的one-hot向量。这对于MSA编码器来说并不难,因为深度神经网络具有极强的拟合能力。然后,我们聚合来自K个one-hot向量的残基i的突变信息。每个向量指示对应同源蛋白质中残基i的背景特异性突变。通过在这些one-hot向量上应用max函数,所有不同的突变信息都将涉及聚合特征。每个0/1聚合特征指示对应的背景特异性突变是否存在于同源物中。然而,在实践中,网络以一种更聪明的方式来探测空间。我们不需要太大的C,因为网络可以将one-hot信息压缩成真实的向量。
2.2.3结构性能预测
为了推断查询蛋白残基的结构性质,我们使用另一个Resnet和一个全连接层,该层以聚合的突变特征为输入,并将结构特性作为最终预测结果。 使用此ResNet的优点是可以捕获残差之间的高阶结构环境。
用于结构特性预测的RESNet的结构与MSA编码器相似。简单地说,该RESNET有8个残差块,每个残差块由两个一维卷积层组成,共64个滤波器。(参照补充图一)我们用不同的随机初始化训练四个相同的Seq-SetNet,然后取这些模型的平均值作为最终的预测结果。
2.3 模型训练过程
2.3.1 输入和输出
Seq-SetNet 将 MSA 作为唯一的输入。在这项研究中,我们通过对查询蛋白质运行三次 HHBlits 迭代来构建 MSA。
HHblits是一项新的蛋白质研究软件,快如闪电且高度敏感,有望显著提高蛋白质的功能性分析
Seq-SetNet报告了总共15个结构特性作为输出,包括表示二级结构的11个特性(包括Q3和Q8标签)和表示扭转角的4个输出。在此,使用DSSP计算二级结构标签和扭转角。
DSSP是用于对蛋白质结构中的氨基酸残基进行二级结构构像分类的标准化算法
为了消除扭转角⨕,⨚的周期性的影响;我们使用它们的正弦和余弦值sin(⨕),cos(⨕),sin(⨚),cos(⨚)代替扭转角。
2.3.2 损失函数
Seq-SetNet中使用的损失函数由六项组成,包括3态和8态二级结构性质的交叉熵损失,以及sin(⨕),cos(⨕),sin(⨚),cos(⨚)的均方误差(MSE)损失。在这个概念验证研究中,所有这些损失项都被赋予相同的权重。
3.实验结果
3.1 数据集和评价标准
我们在两个数据集上评估了Seq-SetNet,即Test2016和Test2018。 Test2016数据集通过运行PISCES获得。
PISCES是一个二维器件模拟软件
它包含12450个具有高分辨结构的蛋白质(R-free<1),用BLAST计算,这些蛋白质的序列同一性小于25%。从Test2016中,我们提取了2015年6月以后沉积的1213个蛋白质作为测试集。 在去除700个以上残基的长蛋白质后,我们将剩余的蛋白质随机分为训练集(10 029个)和验证集(983个)。
使用相同的准则和流水线,我们处理了2018年1月1日至2018年7月16日之间发布在PDB中的结构,得到了另一个测试集Test2018(250个蛋白质)。在Test2018中,与训练蛋白具有>25%序列同一性的蛋白也被过滤掉。 这样,我们保证训练和测试数据集之间没有重叠。
我们评估了 Seq-SetNet 对蛋白质二级结构和扭转角的预测精度,这在以前的研究中得到了广泛的应用。简而言之,对于二级结构预测,我们使用 Q3对3状态标签和 Q8对8状态标签测量预测精度。对于残差的扭转角,我们使用平均绝对误差(MAE)来测量预测角度和真实角度之间的差异。如果误差大于180 °,则由于角度的周期性,将替代地使用解释角。
3.2 蛋白质二级结构和扭转角的预测精度
我们实现了ResNet基线模型,该模型仅采用one-hot氨基酸类型和PSSM作为输入特征(表示为“基线PSSM”)。我们还实现了另一个基线模型,它将HMM配置文件作为额外的特征(表示为“基线PSSM-HMM”)。
Baseline Model是机器学习领域的一个术语,简单来说,就是使用最普遍的情况来做结果预测。
根据表1发现,Seq-SetNet显著优于基线模型。
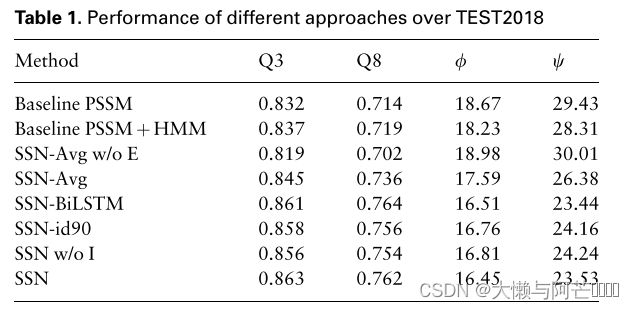
例如,Seq-SetNet模型的3态和8态二级结构的准确率分别为86.3%和76.2%,比PSSM模型的收益率分别提高了3.1%和4.8%。 在基线PSSM~HMM上也可以得到类似的观测。 这些结果清楚地表明,直接利用MSA优于PSSM。
3.3 Seq-SetNet模型各组成部分的贡献分析
3.3.1 Max函数的影响
Max函数是Seq-SetNet的核心部分。如上所述,max函数与深度神经网络一起可以完美地聚合MSA中所有同源蛋白的突变信息。但是平均函数也具有良好的数学性质,可以满足我们的要求:平均函数可以处理可变数量的输入值,并且不受输入值顺序的影响。同时,平均函数在许多高级神经网络中被用作平均池化(He et al.,2016),显示出强大的能力。实际上,平均函数已经在PSSM中使用,其计算每个列的氨基酸分布,并作为几乎所有先进方法中最重要的特征。因此,在Seq-Setnet中,最大函数是否能优于平均函数是一个有趣的问题。为了研究最大值函数在聚合模块中的作用,我们通过用平均函数代替Max函数,建立了一个Seq-SetNet的变体(SSN-AVG)。如表1所示,max函数的性能明显优于平均函数。 以SSN为例,其3态和8态二级结构的准确度分别为86.3%和76.2%,产率分别比SSN~AVG提高1.8%和2.6%。 这些结果清楚地表明MAX函数在聚合突变特征方面的优势。
性能更好的一个可能原因是MAX函数考虑了MSA中不同同源蛋白的权重。 已有研究表明,序列加权有助于蛋白质结构预测。通常,不同的同源蛋白质不应贡献相同的量。Max函数可以通过与MSA编码器一起工作来隐式地学习权重信息,因为它可以通过只保留最大值来忽略无用的信息。
3.3.2 MSA编码器的作用
另一个重要且必要的模块是MSA编码器,它对残留物及其周围环境进行变异编码。 为了直接研究MSA编码器的效果,我们首先禁用MSA编码,从而得到一个称为SSN-AVG W/O E的变体。这个模型中,max函数被替换为average函数,因为它在没有编码模块的情况下不能很好地工作:它将只记录MSA中单个列中是否存在某种突变类型,丢弃任何频率信息。结果表明:SSN-AVG W/O E比基线模型略差。这是合理的,因为将平均函数直接应用于one-hot形式的MSA时,计算结果与PSSM大致相同,但没有考虑背景分布。这清楚地显示了MSA编码器的能力和关注周围突变的重要性。
此外,我们评估MSA编码器与不同数量的残余块。如补充图S2中所示,TEST2018上的8状态二级结构预测的准确性随着残差块的数量而增加。

在MSA编码器中具有可变数量的残差块的TEST 2018上Seq-SetNet的性能。Q8二级结构预测的精度随着残差块的数量而增加。即使使用一个残差块,Seq-SetNet也能达到比基线更高的精度。在达到8之后,精度逐渐平缓。这些结果清楚地表明MSA编码器从MSA中提取特征的优势。
为了显示Seq-SetNet的鲁棒性(模型或者系统的健壮性,受不良影响能否不崩溃),我们用另一种流行的语言模型BiLSTM(双向长短期记忆网络)替换ResNet(残差网络),从而得到一个名为SSN-BiLSTM的变体。具体来说,我们在MSA编码器中使用一个包含96个隐藏单元的三层BILSTM和另一个相同的BILSTM来处理聚集特征和预测结构属性。确定的参数数近似于Seq-SetNet的参数数。BILSTM和ResNet的性能几乎在同一水平,表明Seq-SetNet对超参数不敏感。我们在Seq-SetNet中使用ResNet主要是因为卷积层具有较好的并行能力,这导致了快速的训练和推理过程。
3.3.3 多序列比对的影响
作为一种直接处理MSA数据的方法,MSA的质量对Seq-SetNet的影响比其他方法更大。先前的研究已经表明,预测的质量与Meff高度相关,Meff是MSA中记录的有效同源蛋白的数量。下图是Q8二级结构预测和Meff精度之间的相关性,可以看出,Seq-SetNet的8状态二级结构精度随着Meff而增加。

另外,我们还与PSSM-based方法进行比较。从图可以看出,当Meff的对数<2时,Seq-SetNet的性能处于基线模型的相同水平。这是合理的,因为当没有可用的同源物时,Seq-SetNet将不会从MSA捕获比PSSM更多的突变信息。随着Meff的增加,Seq-SetNet显示出其强大的提取MSA信息的能力,并导致显着提高性能。
此外,一个有趣的观察是,MSA的生成方法导致相当大的影响Seq-SetNet的性能。我们生成了具有99%序列同一性截止值的MSA,而在大多数先前的方法中使用90%同一性截止值。我们训练了Seq-SetNet的另一个变体,其使用90%的同一性截止值来生成用于训练和测试集的MSA,名为SSN-id 90。在表1里,Seq-SetNet(SSN)比变体SSN-id 90的性能稍好。基于PSSM的方法不能从这样一个较弱的截止中表现得很好。一个可能的原因是,90%的身份截断足以生成高质量的PSSM。尽管99%的同源性切断会引入更多的同源蛋白,但这些额外的序列可能不会对PSSM的价值产生太大影响。然而,我们期望从MSA中获得更多的信息,因此我们应该尽可能多地保留同源蛋白。结果清楚地表明,即使大多数同源蛋白具有很高的相似性,SEQ-SETNET也能因为含有了大量同源蛋白而表现得很好。
我们进一步探讨了插入区在MSA中的作用。 由于以往方法的局限性,在计算PSSM时绝对舍弃了插入区域。 在我们的“编码和聚合”策略中,MSA的插入区域信息可以很容易地被MSA编码器捕获。 为了直观地显示插入区域的影响,我们训练了另一种Seq-SetNet,它对所有训练和测试数据都去除MSA的插入区域,称为SSN W/O I。如表1所示,删除插入区域会导致相当大的性能下降。虽然MSA中的插入区通常被认为缺乏信息,但我们的实验表明,这些信息有助于在精心设计的神经网络中预测蛋白质结构。
3.3.4 聚合模块中MAX函数的探索能力分析
max函数相对于平均函数的一个优点是max函数可以从同源蛋白中捕获需要的信息。不同的同源蛋白对最终预测的贡献可能有很大的不同。
为什么Max函数在最终结果预测中发挥了巨大的作用?以蛋白质6bzfB为例:
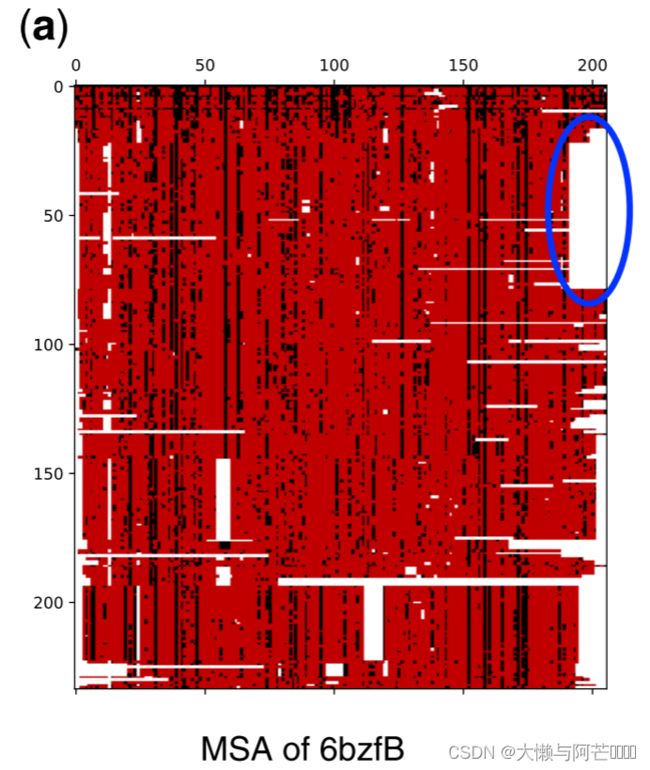
如果同源残基中的至少一个特征被max函数选择为聚集特征,则该残基绘制成黑色。如图所示,几乎所有的同系物都至少有一个有显著贡献的残基,暗示了Seq-SetNet的广泛探索范围。根据HHBlits提供的相似性对同源序列进行排序。相似性较大的同系物通常在黑暗中有较多的残基,表明Seq-SetNet很可能从这些高质量的同系物中获得信息。此外,大间隙区域(蓝圈)对最终预测几乎没有贡献,这表明Seq-Setnet具有滤除噪声的能力,只捕捉必要的信息。
我们进一步研究了有助于聚集特征的同源蛋白的量。我们检查了TEST2018数据集中所有蛋白质MSA的探索程度。在下图中,对于Test2018中的几乎所有蛋白质,Seq-SetNet使用MSA中至少一半的同源蛋白质,对于大约三分之一的蛋白质,Seq-SetNet使用所有同源蛋白质。当MSA具有超过3000个组分同源物时,通常会出现没有贡献的同源物,部分原因是Seq-SetNet使用的特征通道数量有限。当同源蛋白质与查询蛋白相似性较小,它们也可能没有贡献。一个可能的原因是它们在自然界中不是同源蛋白,在寻找MSA时被误选。 聚合模块,特别是max函数,具有滤除此类噪声的能力。

上图表明,当使用最大聚集函数时,TEST 2018数据集中蛋白质的MSA探索范围。这清楚地表明Seq-SetNet可以捕获整个MSA而不是部分同源物的信息。
3.3.5 与现有方法的比较
我们评估了Seq-SetNet,并与流行的二级结构和扭转角预测方法进行了比较,包括DeepCNF、RaptorX-Angle、MUFOLDSS、MUFOLD-Angle、SPOT-1D。为了简洁起见,MUFOLD-SS和MUFOLD-Angle被称为MUFOLD,DeepCNF和RaptorX-Angle被称为RaptorX。我们使用Seq-SetNet的集成版本进行了比较,其中包括四个相同的模型,在训练时具有不同的初始化。

如上图所示,Seq-SetNet在二级结构和扭转角的预测精度方面优于所有三种方法。例如,在TEST 2018数据集上,RaptorX显示Q3为0.816,而Seq-SetNet显示Q3为0.870。此外,Seq-SetNet在⨕(15.90)和⨚(22.31)中显示出比RaptorX(分别为21.01和35.95)更低的预测误差。在TEST 2016上,Seq-SetNet也优于SPOT-1D,即使它使用预测的接触图作为额外特征。对于所有四个预测的性质,观察到TEST 2016和TEST 2018两者的类似性能,表明Seq-SetNet的稳健性。
我们还将Seq-SetNet与其他方法在CASP 13和CASP 14靶点上进行了比较。

可以看出,在32个CASP 13 FM靶标上,Seq-SetNet实现了比SPOT-1D更高的二级结构预测准确度(对于3态和8态二级结构,分别为0.863对0.837,0.762对0.719)。可以在CASP 14靶标上获得类似的观察结果。
MSA可能具有成千上万的同源蛋白质,而PSSM的大小是固定的,并且仅由靶蛋白质长度决定。研究Seq-Setnet是否可以在合理的时间内完成预测是有趣的。由于使用的参数数量少(<1 M)和高效的实现,即使直接处理MSA,Seq-SetNet也可以在普通计算机上在合理的时间内完成预测。以CASP 13中的T1000-D2蛋白为例,该蛋白有431个氨基酸残基,MSA中有3973个同源蛋白,即使使用Intel CPU 2.8 GHz,Seq-SetNet也能在25 s内完成整个预测过程。
4. 总结
这里提出的蛋白质二级结构和扭转角预测的结果突出了直接使用MSA作为序列集的特殊功能。Seq-SetNet利用深度网络可以从复杂的MSA数据中自动提取相关特征的能力,而无需任何手工特征。
虽然在概念验证研究中,我们展示了Seq-SetNet在预测残基的二级结构和扭转角中的应用,但该概念和基本思想可以扩展到阐明残基-残基接触和距离。在未来的研究中,这将被纳入Seq-SetNet,这将极大地促进蛋白质结构预测。
5. 补充
使用seq-setnet预测片段结构的例子。 (a)5YO8A:残基11-50,RMSD=1.73°A; (b)6CVZB:残基76-115,RMSD=3.07°A; ©5Z8OB:残基2-41,RMSD=2.07°A; (d)5UB3A:残留物68-107,RMSD=2.01°A。 预测的结构用红色表示,本地结构用绿色表示。
