分子结构的子结构查询:SMARTS语言
在化学信息学中,有时候我们要确定化学分子是否包含特定的模式(结子构)
之前我们已经提及,fingerprint的SMILES字符串经常用来表示分子。
SMARTS是SMILES语言的扩展,可以用于创建查询,类似文本语言的正则表达式。
任何的SMILES字符串都可以是SMARTS字符串
下面展示,如何从SMILES字符串定义分子,显示分子,并突出与特定SMARTS,模式匹配的原子
1. 导入基础模块
from rdkit import Chem
from rdkit.Chem.Draw import MolsToGridImage
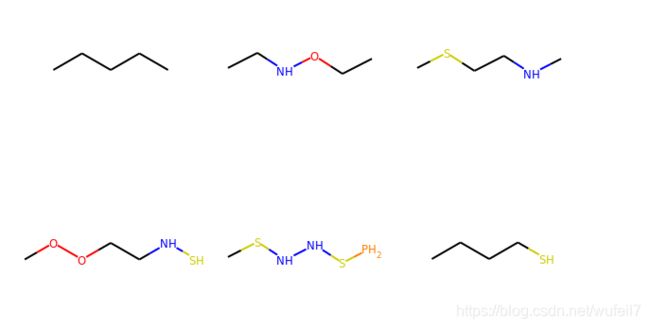
2. 定义化学分子
smiles_list= ['CCCCC', 'CCNOCC', 'CSCCNC', 'COOCCNS', 'CSNNSP', 'CCCCS']
mol_list = [Chem.MolFromSmiles(x) for x in smiles_list]
3. 分子结构可视化
MolsToGridImage(mol_list)
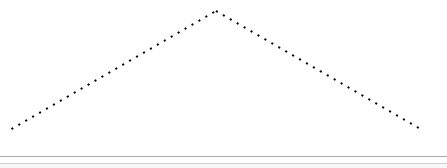
4. 建立一个查询请求
查找有三个C原子组成的脂肪链
query = Chem.MolFromSmarts('CCC')
query
5. 查找匹配
match_list = [mol.GetSubstructMatch(query) for mol in mol_list]
match_list
输出:[(0, 1, 2), (), (), (), (), (0, 1, 2)], 代表第1个mol对象(’CCCCC‘)分子,有三个部分与query相同;第6个’CCCCS’分子,有三个部分与query相似。查询结果确实符合我们的query.
6. 可视化
注意这里高亮仅仅显示了第一个匹配的部分。
MolsToGridImage(mols = mol_list, highlightAtomLists=match_list, molsPerRow=4)

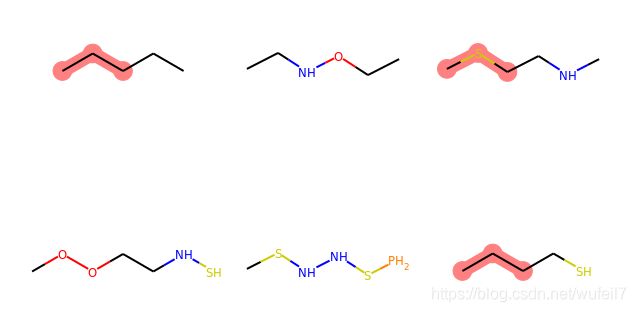
7. 匹配特定模式的分子
SMARTS中,*可以代替任何的原子,例如:
query = Chem.MolFromSmarts('C*C')
match_list = [mol.GetSubstructMatch(query) for mol in mol_list]
print(match_list)
MolsToGridImage(mols=mol_list, highlightAtomLists=match_list, molsPerRow=3)
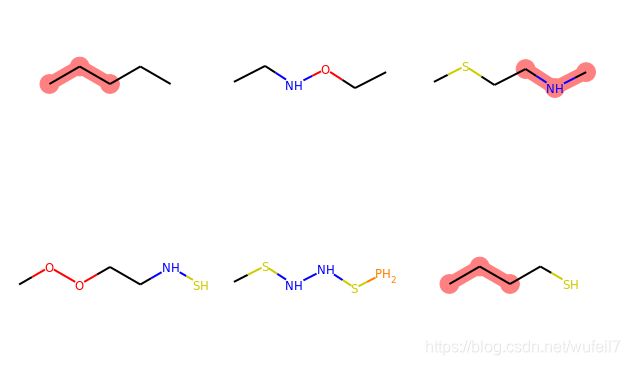
SMARTS中,[]代表中括号中间的一个原子可以是N,也可以是C,例如:
query = Chem.MolFromSmarts('C[N,C]C')
match_list = [mol.GetSubstructMatch(query) for mol in mol_list]
print(match_list)
MolsToGridImage(mols=mol_list, highlightAtomLists=match_list, molsPerRow=3)
8. 备注
SMARTS还可以有其他复杂的功能,需要去google一下。