scRNA-seq Course 学习
对scRNA-seq的介绍
scRNA-seq是对单个细胞进行测序分析,便于研究单个细胞水平的问题,例如:鉴定细胞类型,鉴定细胞基因表达差异,细胞反应的差异,单个细胞内部的基因表达调控。
scRNA-seq和bulk RNA-seq相似,区别只在于研究的细胞数量,后者是对同一组织的大量细胞进行转录组分析,而前者是对单个细胞进行转录组分析。
scRNA-seq的流程包括:实验流程,原始数据处理,数据分析获得生物学意义。
目前也有不同的测序方法:SMART-seq2, CELL-seq,Drop-seq。
目前也有很多商业测序平台:Fluidigm C1, Wafergen ICELL8,10XGenomics Chromium。
实验流程
scRNA-seq的实验流程包括:从组织里分离出单个细胞,提取RNA,逆转录和二链合成生成cDNA, PCR扩增cDNA, 构建测序的文库,测序,获得单细胞表达谱,鉴定细胞类型。

该图为单细胞测序的实验和分析流程。IVT是in vitro transcription, 体外转录。
目前也有一些平台搭建了测序数据分析流程:Falco, SCONE, Seurat(R包,用于QC,analysis), ASAP(Automated Single-cell Analysis Pipeline)。
目前在单细胞测序领域很大的挑战是:PCR扩增倍数, 基因信号丢失。
目前也有很多单细胞测序的实验方法,方法的关键在于基因计数和捕获单细胞效率。
计数:目前存在两种类型,即全长(Full-length)和标签计数(Tag-based)。
基于转录本全长的计数要求对全长的序列达到一致的覆盖度,然而,测序本身就对序列3’段存在偏好性。
基于标签计数,也就是只捕获3‘端的RNA序列或5’端的RNA序列,这个计数方法可以与UMI连用提高计数的效果。但是,由于仅对3‘端或5’端计数,可能会减少序列的比对效果,不利于区分不同序列。
捕获单细胞:目前捕获单细胞的策略影响了测序平台的通量。目前广泛使用的捕获单细胞方法有:mircowell-based、microfluidic-based, droplet-based,分别把细胞置于微孔板上,微流体,微滴中。
在基于微孔板的平台上,细胞通过移液枪或激光捕获进行分离并置于微孔板中。这个方法的优点是可以和流式细胞术结合,基于细胞表面分子分子筛选细胞,另一个优点是可以给细胞拍照。缺点是这个方法通常是低通量的,每个细胞需要的工作量很大。

在基于微流体的平台上,捕获细胞和文库制备过程都整合在同一张芯片上,通量较高,但是只有接近10%的细胞能够被捕获,因此,该方法并不适用于稀少的细胞,芯片很贵,但能够节省用于反应的试剂费用。

在基于微滴的平台上,用纳升的微滴将单个细胞和磁珠包裹起来,这个磁珠上面有用于构建文库的酶,特定的barcode。所有微滴都可以被混合,同时测序。基于微滴的测序平台有很高的通量,每个细胞的测序费用在0.05美元(USD)。然而,测序的费用依然限制了测序结果,测序覆盖度低,仅有几千个不同的转录本被检测。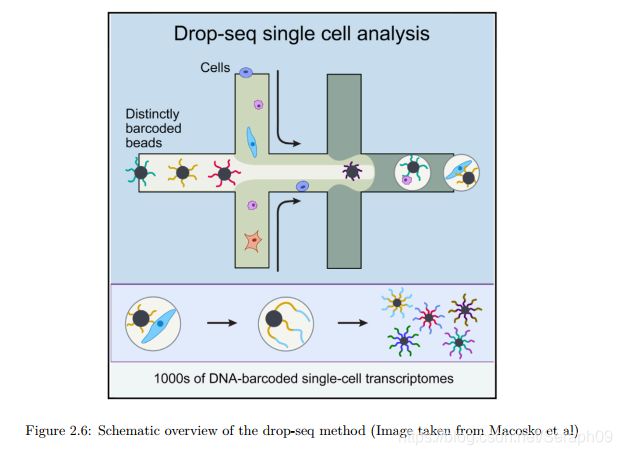
数据分析
scRNA-seq的数据分析流程包括:
- 序列质控(FASTQC)
- trimming去除adapter
- 序列比对生成BAM文件
通过FASTQC,可以知道测序的序列质量。FASTQC后会生成一个.zip文件和一个.html文件,可以通过filezilla或者scp将结果下载下来。
通过trimming(修剪)可以去除序列的adapters(接头)和低质量的序列。
利用trim_galore可以去除有问题的adapters(通过FastQC report “Adapter Content”图查看adapter的情况)。对序列进行trimming后再用FASTQC产生另一个报告进行比对。
测序文件的类型:
从测序平台下载下来的原始数据为FastQ数据。对于单细胞测序来说,所有的测序方法都是采用双端测序。
根据测序的方法,Barcode序列可能或出现在一条或单条reads上。
根据实验方法,利用UMI(Unique modified identifier)的实验技术,序列通常会包括adapters, UMI barcodes和cell barcodes。
FastQ 文件的数据格式:

将FastQ文件进行samtools比对后获得BAM文件,BAM文件存储比对的序列信息。一些仪器会将FastQ序列直接比对到一个标准基因组上,生成BAM或者CRAM文件。然而,这样的比对并没有包括外源的参考RNA序列ERCC(External RNA Controls Consortium),因此ERCC序列便不会生成BAM/CRAM文件。为了保证能比对到多个位置的序列最终结果只有一条序列上,可以用samtools去除除第一条以外的其它序列。CRAM文件和BAM序列相似。
用于比对的参考序列是基因组序列(FASTA)以及注释文件(GTF),这些文件可以来源于Ensembl, NCBI, UCSC Genome Browser。
来源于不同版本的基因组中,Ensembl版本是最易使用且含有大量的注释信息,NCBI的注释非常严格,注释可信度高,UCSC版本含有运用不同标准的大量基因注释信息。
测序数据的分解
测序原始数据分解是否需要使用,依赖于实验方法。
目前最灵活的用于分解测序原始数据的流程是zUMIs。
对于某些测序方法,返回的原始数据已经进行分解,例如:Smartseq2 或者一些双端测序方法。
在公共数据库GEO和ArrayExpress中,小规模数据或基于微孔板测序的数据需分解后才能上传,而且多数测序仪器都会自动分解数据再返回给用户。
如果使用的不是公共流程,原始数据并没有被分解,则需要用户自己来分解原始数据。
分解数据需要确定并移除序列的cell-barcodes(如果有UMI,也需要分解UMI值)。分解数据前,应该提前知道数据的cell-barcodes。而且,需对原始数据中的cell-barcodes与预期的cell-barcodes进行比较,因为,由于cell-barcodes本身的设计,会存在部分barcodes的错配。
对于包括UMI的测序原始数据,分解时应将UMI的代码放在基因转录本的名称前面。对于Barcode数远大于细胞数,那么barcode也会被放在基因转录本名称前。
基于微滴(droplet-based)的实验方法中,仅有部分微滴含有磁珠和完整的细胞。一些从死亡破碎的细胞中产生的RNA序列也落入微滴中,经过扩增,测序,成为了单细胞测序的背景噪音。
为了去除这一来源的背景,使用多种方法。
例如:通过对每个cell-barcode对应的转录本分子进行统计,试图找到大文库和小文库序列之间的"break point", 并将小文库样本视为背景处理,即找到每个cell-barcode下文库大小的变化的突变值(可进行log转化或不进行转化)。
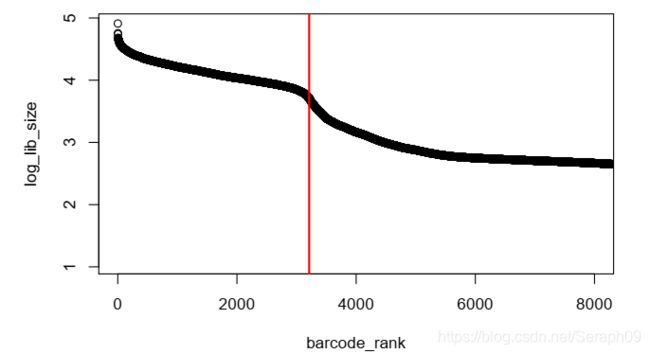
或者是将每个样本的数据分布情况进行整合,找到低分布和高分布数据之间的交叉点。
序列的比对
将原始数据序列进行质量控制,trim adapter以及 demultiplex cell barcodes, UMI后,就要将原始数据和参考基因组进行比对。
分解好的测序数据,可以用STAR或者Kallisto去比对序列,序列比对的策略包括两种,STAR的sequence alignment 以及Kallisto的Pseudo-Alignment
利用STAR进行序列比对,也就是常规的reads mapping 到参考基因组上。
STAR会尽可能找到比对到基因组上的最长的序列,也被称之为"splice aware"的对比方法,也就是说STAR比对序列的时候,能够对应识别RNA的剪接事件,跨外显子查找序列。
通常STAR比对,能够发现RNA上新的剪接事件,但运行该程序需要很大的内存。
运行STAR的时候,用户首先需要提供参考基因组序列和注释序列,STAR生成基因组索引。然后,STAR才会将用户的reads数比对到基因组索引上。

该图表明RNA-seq的序列部分比对到基因组上的外显子区域,另一部分再截取出来,比对到基因的另一个外显子区域。
另一种比对方法是Kallisto的伪比对方法(Pseudo-Alignment),和STAR的比对方法的区别在于Kallisto的伪比对是将k-mers比对到参考基因组上。
k-mers是reads转化而成,也就是reads转化成k-mers,k可以为5,6,7等,分别对应长度为5,6,7的k-mers。

该图表明为一条序列能够生成的k-mers
Kallisto Pseudo-aligner相较于STAR的比对,速度更快,同时在某些情况下,Kallisto pseudo-aligner的方法更能够适应测序错误,原因在于第一个k-mer第一个碱基出现的测序错误,并不会在第二个k-mer上出现。
其次, Kallisto Pseudo-aligner比对的参考基因是参考转录组,这表明Kallisto Pseudo-aligner比对的是RNA异构剪接体。
然而,scRNA-seq本身测序覆盖率低于bulk RNA-seq, 因此用于比对的有用测序信息就会减少。
许多scRNA-seq方法具有3’-端的偏好性,这也意味着当两个isoform的转录本的区别发生在5’端,则不能清楚知道序列来源于哪一个isoform转录本。
部分scRNA-seq的流程中,转录本的序列长度很短,则更不能清楚地知道序列来源的isoform。
Kallisto的pseudo alignment比对时,会将序列比对到一个等价的转录本聚集体上,这意味着如果一条序列比对到多个转录本上,Kallisto会记录比对到多个转录本的情况。这多个转录本形成的聚集体,也就是equivanlance class。
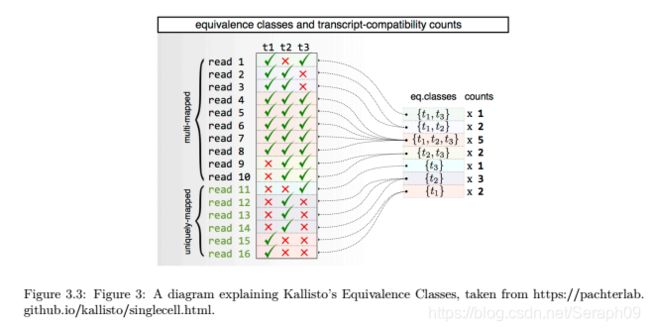
该图表明Kallisto的Equivalance Classes的集合
Kallisto Pseudo-Alignment比对的输出结果是matrix.cells, matrix.ec,matrix.tsv和run_info.json。
matrix.cells:包含的内容是细胞的IDs信息
matrix.ec:包含的是equivalence class的信息。
matrix.tsv: 包含的信息是每一个细胞有多少的reads比对到equivanlance class中。
run_info.jason:包含的信息是关于Kallisto是如何运行的。该信息可以忽略。
构建基因表达矩阵
比对后需构建作为分析基础的基因表达矩阵,基因表达矩阵的构成:列为细胞名称,行为比对的基因,每一个值代表在该细胞下该基因的表达水平。
序列质控和比对
构建基因表达矩阵的来源是比对后的bam文件。
在构建基因表达矩阵时,仍然需要对原始数据序列的质控,也就是Reads QC,可以用Integrative Genomics Browser (IGV)或 SeqMonk查看序列质量情况。
去掉低质量的序列以后将剩余的序列比对到基因组去。常用的工具是STAR或者TopHat。
值得注意的是:当加入了内参(spike-ins)的时候,需要在比对前将内参序列加入参考基因组中。而当测序序列中有UMI的时候,就需要将UMI的Barcode从序列中删除。
在比对中,我们应当保证每个细胞有60%-70%的序列能够比对到参考基因组中。低比例的比对序列通常表明是序列的污染。
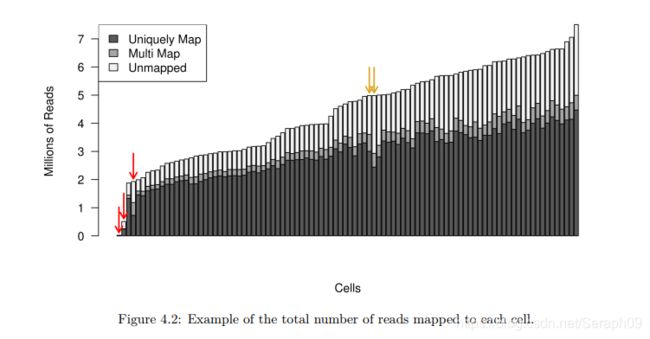
图示为:在一个单细胞测序实验中对所有细胞的序列比对情况分析,红色箭头表明序列少且对唯一比对到的序列少,黄色箭头表明没有比对到的和多比对序列多
比对结果质控
对序列进行比对后,仍然需要对比对质量进行评价。
评价的方法包括:比对到tRNA/tRNAs的序列数量,唯一比对到的序列占比,比对到剪接体连接位置的序列以及比对到转录本的序列深度。
在bulk-RNA seq中对比对结果进行评价常用RSeQC, 这个软件也可应用到scRNA-seq上。
比对序列定量
然后是对序列进行定量,对于mRNA数据,我们可以使用用于bulk RNA-seq的工具:HT-seq 或者 FeatureCounts。
针对某些UMI的测序平台,定量方式有所不同。
由于单细胞测序有UMI(Unique Molecular Identifiers,唯一分子标记)(这个分子标记是理论上唯一标记细胞单个转录分子的标记,可用来消除PCR扩增噪音以及scRNA seq的偏好性),因此计数上需要对UMI进行考虑。

这个图表明一个转录分子上的结构是包括PolyA尾巴,UMI和细胞的Barcode.
R/Bioconductor
对比对序列进行定量后,需要利用R/Bioconductor进行后续的序列分析
Tabula Muris
Tabula Muris 是一个整合单细胞测序数据的组织。
清理基因表达矩阵数据
获得基因表达矩阵以后,对于基因表达矩阵仍要进行数据清理。
对于基因表达矩阵的数据清理的难处在于目前质控的方式多种多样,因此不能以质控后获得的数值作为评价,可以选用计数结果中的异常值作为质控的评价指标。
对测序的细胞进行质控可以从以下三个方面:构建的文库大小,检测到的特有基因数量,以及ERCCs与内源性的RNAs比例评价细胞的测序质量。
构建的文库大小以读取的counts数作为指标,文库过小意味着细胞破碎或者微孔板并没有成功捕获到细胞。
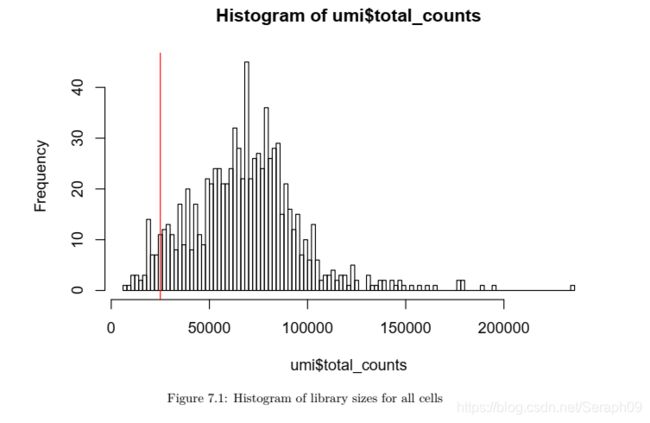
该图的横坐标为每个细胞的文库大小,纵坐标为对应细胞的个数。
每个细胞样本检测到特有基因数应当在7000以上(对于具有高深度的RNA测序结果而言,真实数据检测到绝大多数样本检测到的基因数为7000-10000之间),因此独特基因数低于7000的细胞样本就会被排除。
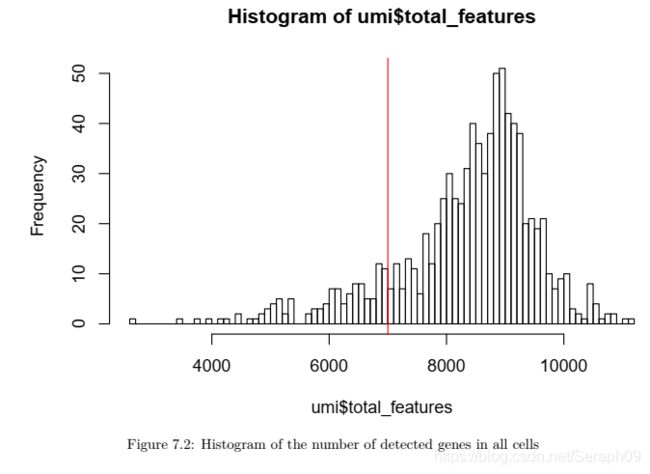
该图表明每个样本可以检测到的独特基因数量,纵坐标为样本个数。
向细胞样本中加入ERCC,通过衡量ERCC和内源性RNA的比例,可以估计捕获细胞的RNA总量。假如细胞中ERCC的比例过高,则意味着细胞内源性的RNA很少,则意味着该细胞死亡或破碎并导致RNA的降解。

该图表明为不同样本中ERCC的比例情况,横坐标为检测的基因数,纵坐标为ERCC的counts比例。
对基因表达矩阵的细胞进行质控以后,就要对细胞进行过滤。
过滤的方法有手动过滤和自动过滤。
手动过滤根据细胞质控的结果进行分析,自动过滤也就是通过对一系列QC矩阵进行PCA分析找出异常值(outliers)进而确定存在问题的细胞。
如下图矩阵。

该图中feature应该是检测的基因数。
scater包分析异常值的时候,会提前生成 行为细胞,列为不同QC结果值的矩阵,并进行PCA的分析。

该图表明检测细胞异常值的PCA图。
除了对细胞的质控分析以外,还有对基因的质控分析。
通常而言,我们怀疑由于技术原因导致基因表达异常,基因表达前50的基因可能并不是正常的基因表达结果。

我们也会移除基因表达水平被定义为“不可检测(undetectable)”(利用UMI计数中,一个基因detectable意味着至少两个细胞中含有至少一个该基因的转录本。而利用reads计数,则意味着至少在两个细胞中含有超过5条该基因的reads。)
通过对基因表达矩阵的细胞和基因进行质控后,我们终于获得了经过质控后数据。
基因表达数据的可视化
通过对基因表达矩阵质控后,获得质控后数据。
对于单细胞测序数据,我们仍然需注意不应将批次效应产生的差异作为细胞本身的差异。
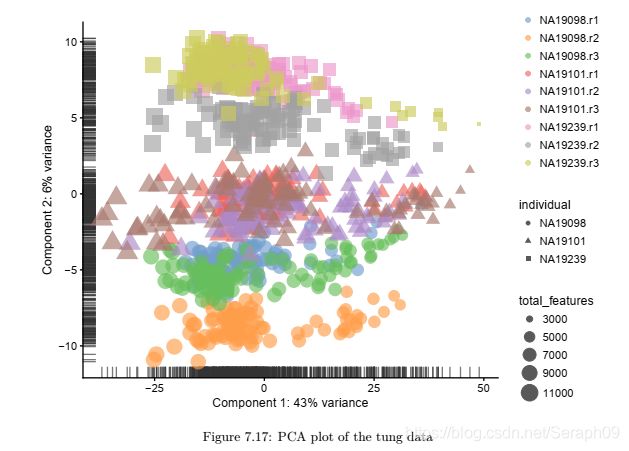
通过降维的方式对数据进行可视化,例如:PCA(主成分分析),tSNE(t-分布随机邻近嵌入),从而对所有的基因进行降维并作图。
PCA降维是将多种细胞进行分类的相关变量转换成主成分(包含多个不相关的特征值),主成分2则为次相关的变量。从而对细胞降维并投射到以PCA1和PCA2为坐标轴的二维空间。
PCA的主成分与表达矩阵的协方差矩阵的特征值相关,这里涉及数学方面的内容,不甚了解。

PCA

【在QC之前应该对基因的counts数进行log转化,这可以使基因的表达偏向正态化,同时有利于减少主成分分析中对主成分的变动。】
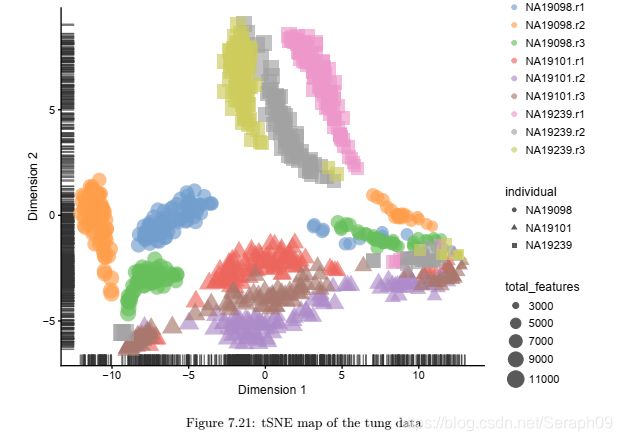
tSNE降维则是将降维和邻近网络结合用于分析高维数据。由于tSNE使随机算法,这意味着对同一个数据使用该算法会产生不同的图片。为了避免这种情况的发生,我们可以设定seed值。
tSNE降维需要提供混乱度(perplexity),用于构邻近网络。高值则在降维后细胞十分密集,低值则在降维后细胞比较稀疏。
scater采取的perplexity默认值为:总细胞数量/5

log转化以后需要进行标准化,根据细胞的文库大小进行标准化。
确定导致基因差异的混淆因素
scRNA-seq在技术上很难构建真正的生物学重复,猜测原因在于相同样本,捕获的细胞,扩增的效率的变化都会很大。因此,对基因表达矩阵中不同细胞不同基因的counts数量的差异可能来源于人为因素,我们需要去除来源于实验或技术本身的差异而导致的差异。
来源于技术本身的原因有:
检测的基因数,测序深度,ERCC占比,批次效应,实验方法本身的差异(测序偏好A/T碱基,捕获短序列转录本的能力)。
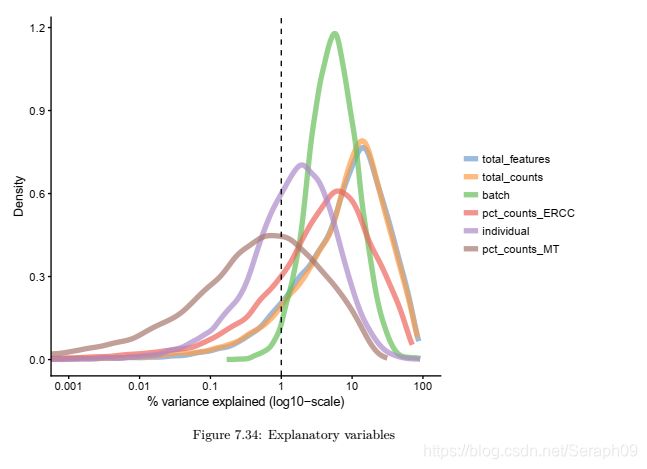
去除混淆因素可以通过加入spike-ins共同测序。
spike-ins用于测序,从而构建spike-ins 与 endogenous genes的模型,从而推断技术导致差异的混淆因素。
spike-ins的方法在BASiCs, scLVM, RUVg的平台使用。但是spike-ins存在的问题在于spike-ins 的序列数量可能比内源性基因的还高。
去除不需要的变量,方法多种多样,例如:RUVseq(Remove Unwanted Variation),
RUVseq可以删除掉不需要因变量元素,包括RUVg, RUVs。
如果实验设计好,可以使用Combat减弱批次效应。
mnnCorrect也可以减弱批次效应。
GLM是Combat的简单线性回归模型。
如何评估比较不同去除confounder(混合因素)策略的效率。评估的难点在于不知道哪些是技术导致的混淆因素,哪些是生物学差异。
下面的内容不是很清楚,因为不理解。
评价效果1: 评估标准化用PCA
评价效果2: 评估正确率用RLE
评价效果3: 评估批次效应是否去除
评价效果4:评估批次效应是否去除
kBET利用KNN网络检测批次效用
标准化
确定导致差异的混淆因素之外,需要对基因表达矩阵进行标准化,以去除由于混淆因素导致的差异。
最主要的标准化是利用文库大小进行的size-factor标准化。
不同平台测序得出的library size(文库大小)是不同的,一些定量方法结合文库大小定量,不需要进行文库大小的标准化,例如:Cufflinks,RSEM。其它的方法则需要根据文库大小进行标准化。
常用于bulk RNA-seq的标准化也可以应用于scRNA-seq,例如:UQ,SF,CPM,RPKM,FPKM, TPM.
CPM: 这是最简单的标准化,计数为每百万counts的计数。也就是假设每个细胞有100万的文库大小,计算每个细胞的不同基因有多少的
counts。该标准化方法的潜在缺点在于如果存在高表达基因,在所有细胞中都表达,同时具有差异,那么统一文库大小,可能会忽略该高表达基因的差异。
RLE(size factor, SF): 大小因子。由于计算的是几何平均数,因此不适用于表达为0的基因,也不适用于测序深度低的scRNA-seq。
UQ(upperquartile):上四分位数。标化因子是(各样本的上四分位数对应counts数)/(各样本上四位数对应counts数的中位数)

该图表明大致的标化情况
TMM:该方法对M-values值进行加权裁剪获得均值。
scran: scran是个R包,利用类似CPM的方法进行标准化。
Downsampling: 这种方法是对基因表达矩阵随机取样,但是取样数小,最后保证每个细胞有接近相同的counts总数。但是由于随机取样的不确定性,每次取样的并不相同。因此需要多次进行取样,确保结果稳定。
值得注意的是,用于bulk RNA-seq的标准化方法中,RLE,TMM,UQ可能并不适用于scRNA-seq,原因在于这些标准化方法的一句是具体的实验情况。
除了对counts数进行标准化以外,也有对基因/转录本的长度进行标准化的方法,例如:RPKM,FPKM,TPM.
RPKM: Reads Per Kilobase Million,每百万reads,千个碱基对应的数量。这个方法用于单端测序。
FPKM: Fragments Per Kilobase Million ,每百万reads,千个碱基对应的数量。这个方法适用于双端测序,同时两条序列对应于同一个fragment。
TPM: Transcripts Per Kilobase Million,每百万千各碱基序列对应的数量。这个方法先均一化基因长度,在均一化reads数。
以上对长度进行均一化的方法应用于bulk-RNA seq,但对于scRNA seq而言,对长度进行均一化的方法并不适用,因为测序的时候,对UMI(Unique molecular identifier)进行率先测序了的。
生物学意义分析
对基因表达矩阵进行标准化,去除混淆因素后,我们便需要分析差异相关的生物学因素了。
scRNA-seq生物学分析常包括基于转录本注释细胞类型,
关于确定细胞类型,问题在于没有监督聚类,因此,需要根据和转录组相似性进行聚类分析,然而,在大多数情况下,我们甚至事先不知道聚类的数量。
对于大量数据分析前,我们最好先降维分析。
需要质控后的基因表达矩阵进行无监督聚类分析。常用的聚类方法有:hierarchical clustering, k-means clusting, graph-based clustering。
hierarchical cluster:层级聚类的方法,包括从下至上层级聚类和从上至下聚类。
从下至上的层级聚类也就是原细胞样本先聚类成簇,然后不同簇再聚类,逐级向上。
从上至下,则是从集群逐渐拆分向下,形成层次结构。
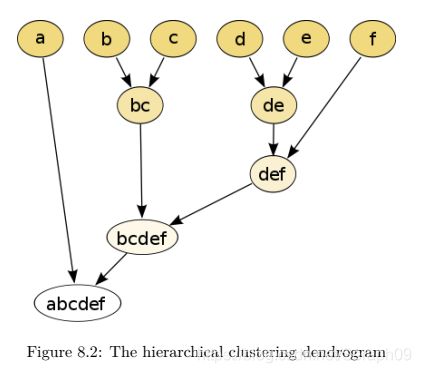
k-means: 提前确定细胞类型数量,进而确定存在的细胞簇,逐渐迭代,不同细胞就分配到最接近的簇。
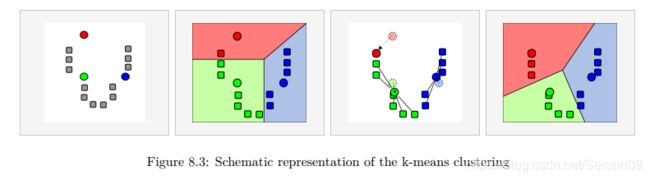
graph-based cluster: 细胞作为图形的结点,构建图形并确定图形边的权重。
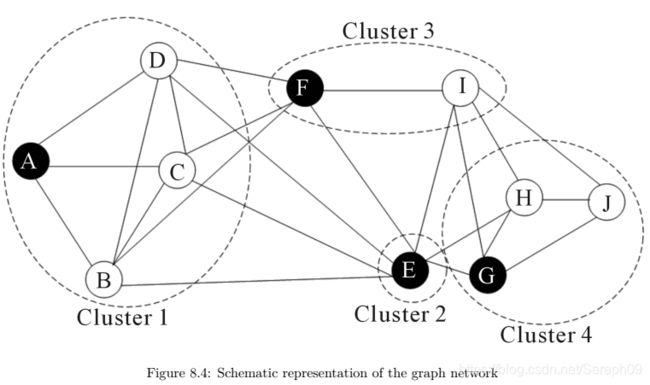
目前单细胞聚类分析存在的问题在于:
- 不清楚聚类数量
- 不知道细胞类型
- 可扩展性
- 工具对于用户并不友好
目前根据降维和聚类开发出多种用于分析单细胞细胞类型的软件,例如:SINCERA, pcaReduce,SC3,tSNE + k-means,SNN-Clip, Seurat clustering。
SINCERA:基于层级聚类,数据需要进行z-scores转化,通过寻找层级结构中的第一个聚类确定簇的个数。
pcaReduce:基于PCA,k-means和迭代的层级聚类方法。
SC3: 基于PCA,降维,k-means,共识聚类。
tSNE + K-means: 基于tSNE, k-means。
SNN-Clip:基于Graph-based的聚类,SNN(Shared Nearest Neighbours)。
Seurat clustering: 类似于SNN-Clip方法。
比较不同的聚类簇,可以使用adjusted Rand index,这用来衡量两个聚类簇的相似度。
特征基因筛选
因为单细胞测序的基因,仅有一部分基因与生物状态相关,例如细胞分化,细胞对环境刺激的反应。scRNA-seq实验检测到的基因受技术噪音的影响而不同,找到特征基因则需要去除技术噪音。
因此,在寻找特征相关信息之前应该去除由于技术噪音产生的基因。
在筛选Feature的时候,我们采用无监督的方法筛选,也就是没有相关的标签(cell-type labels 或者 biological group)。
筛选特征基因需要去掉仅描述数据中技术噪音的空模型,筛选基因表达与空模型不相同的基因。假如数据包含了spike-ins, 可以将spike-ins用于模拟技术噪音。但是scRNA-seq实验中,spike-ins的内参数量很少,模拟技术造影并不好。
对特征基因的定义不同,也会出现不同描述特征基因的方法,特征基因是能够表达出细胞状态不同的基因。
筛选特征基因存在以下5种方法:
方法1:可以把特征基因定义为高度变化的基因(Highly Variable Genes, HVG)。
HGV确定的前提在于所有细胞中表达差异性很大的基因来源于生物学原因,而不是技术噪音。由于基因平均表达情况和the variance in the read counts呈正相关,这有助于合理确定HGVs。

该图显示了平均基因表达情况和基因方差之间的关系,由此可见,基因平均表达水平越高,则方差越大。
BrenneckeGetVariableGenes的方法可以纠正由于基因平均表达情况差异而引起的基因方差的改变,并找到高度变化基因(HVG)。
方法2:特征基因是高信号丢失的基因。
该特征基因可通过M3DropFeatureSelection()函数获得
M3Drop_genes <- M3DropFeatureSelection(
expr_matrix,
mt_method = "fdr",
mt_threshold = 0.01
)
M3Drop_genes <- M3Drop_genes$Gene
方法3:通过DANB()
方法4:特征基因是在某一类细胞类型,与细胞类型成正相关,而在另一类细胞中与细胞类型成负相关的基因。
该特征基因可以通过矩阵相关性分析
cor_mat <- cor(t(expr_matrix), method = "spearman") # Gene-gene correlations
diag(cor_mat) <- rep(0, times = nrow(expr_matrix))
score <- apply(cor_mat, 1, function(x) {max(abs(x))}) #Correlation of highest magnitude
names(score) <- rownames(expr_matrix);
score <- score[order(-score)]
Cor_genes <- names(score[1:1500])
方法5:特征基因是一类主成分比例很高的基因,这更有可能成为特征基因。
# PCA is typically performed on log-transformed expression data
pca <- prcomp(log(expr_matrix + 1) / log(2))
# plot projection
plot(
pca$rotation[,1],
pca$rotation[,2],
pch = 16,
col = cell_colors[as.factor(celltype_labs)]
)
伪时序分析
单细胞测序分析可以研究细胞连续变化的过程,例如一个细胞分化成另一个细胞类型。由于单个细胞每次进行测序都需要被裂解,因此,我们对于某个细胞的研究就需要选择多个时间点的细胞,然后获得该细胞的表达谱,并用统计的方法获得细胞潜在的时间发展轨迹。
目前,用于伪时序分析的工具包括:Monocle,TSCAN, destiny, SLICER和ouija用于对细胞进行排序。
包括:
SCUBA(Matlab implementation)Wanderlust(Matlab and requires registration to even download)
Wishbone(Python)
SLICER(R, package on Github)
SCOUP( C++ command line tool)
Waterfall (R script)
Mpath(R package)
Monocle(Bioconductor package)TSCAN(Bioconductor package)。
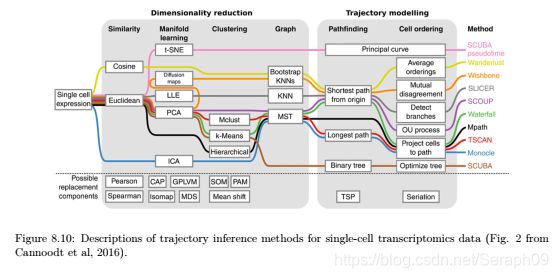
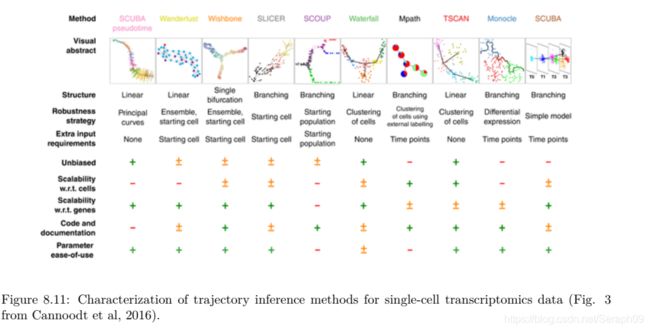
TSCAN(Bioconductor package):TSCAN结合聚类和时序分析。首先利用mclust将细胞进行聚类,然后构建最小扩展树(minimum spanning tree)将聚类结果连接起来。树的分支连接多个聚类作为主要的分支,并用于确定伪时间。
monocle:monocle去掉了TSCAN的聚类,直接构建了最小扩展树。monocle确定了树的最长路径用作确定伪时间。如果出现分叉的路径,则定义为不同的细胞状态。但monocle构建不能使用全部的基因,因此需要进行基因特征筛选(Feature Selection: M3Drop)
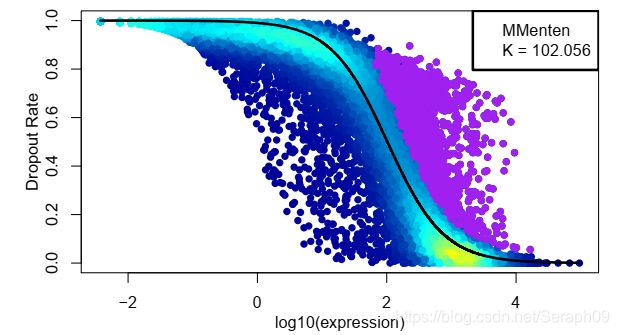
该图显示通过M3Drop进行特征基因筛选的结果。
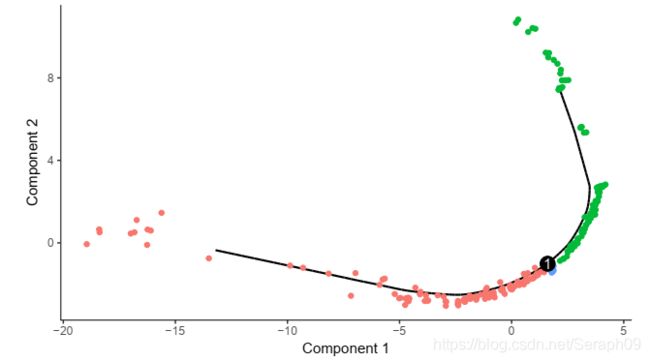
该图是利用monocle进行时序分析。
Diffusion maps:扩散映射,这个方法潜在在于样本来源的数据是扩散过程。Haghverdi等人利用该方法生成了destiny的R包。
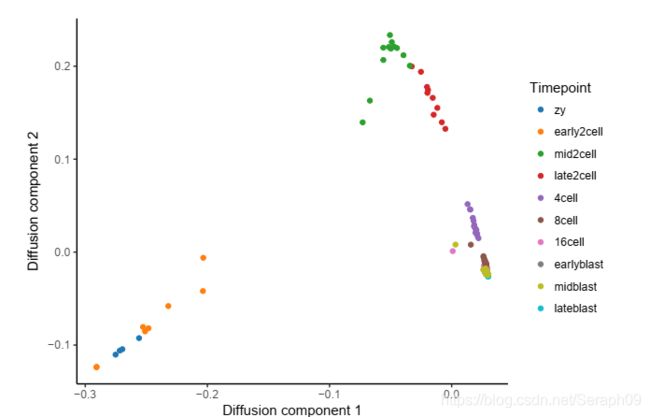
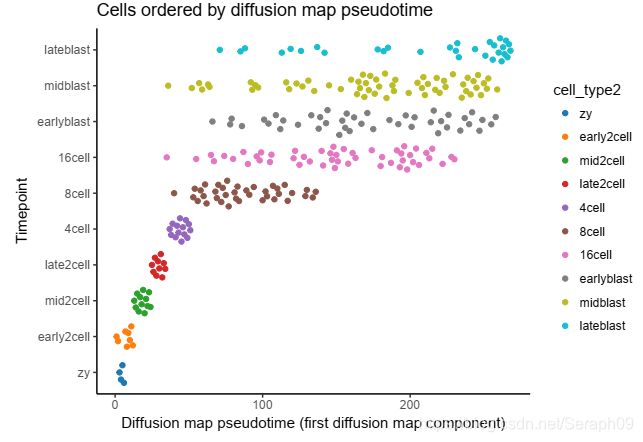
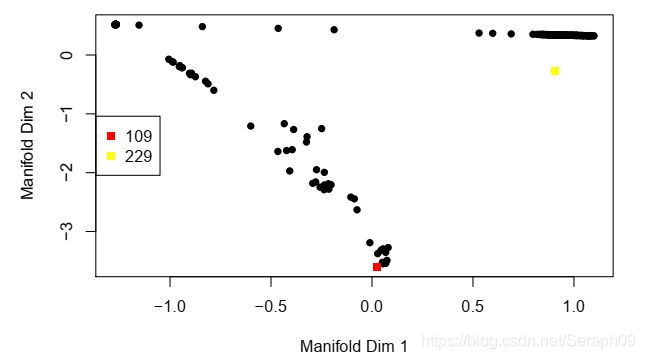
SLICER: SLICER也是通过描述基因表达变化进而构建一系列生物学过程的时序轨迹。SLICER捕获高度非线性的基因变化,自动筛选与过程相关的基因,以及检测多个分支相关特征。

Ouija: 该方法用一些标记基因作为小panel, 进而解释学习单细胞的伪时间。
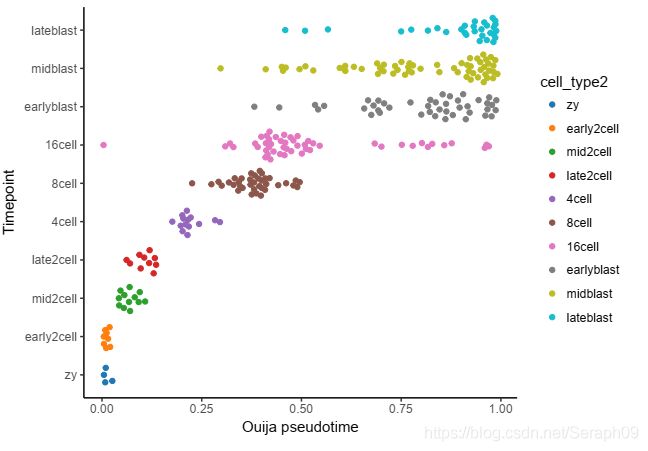 关于上述多种伪时序分析方法,需要进行对比评价。
关于上述多种伪时序分析方法,需要进行对比评价。
基因表达矩阵信号丢失的原因分析(Imputation)
单细胞测序仍然存在的挑战在于检测基因值为0(或许可称之为信号丢失)。
检测值为0的可能性:
- 该基因在细胞内确实没有表达,因此也就没有转录本。
- 该基因确实表达了,但是转录本在测序前丢失了。
- 该基因表达了,而且细胞也被捕获,转录本确实转成了cDNA,但是测序深度不够产生reads。
如果信号的丢失来源于实验因素,我们或许该采取相关的计算机手段进行校正。能够进行基因表达值丢失得归因,则意味着有一个潜在的模型。但是,我们并不清楚信号丢失的原因是技术原因还是本来就没有转录本,那么,对信号丢失的归因就存在很大问题。
目前对于信号丢失归因的方法主要有两种:MAGIC和scImpute。
scImpute: 目前我们可以通过PCA比对原始数据的结果,以及SC3对归因矩阵进行聚类。
MAGIC: 该程序只能运行于Python和Matlab上。
差异基因的分析(DE Differential Expression analysis)
差异基因是同一基因在不同环境,时间,空间,压力下,转录表达的水平出现差异或是发生基因突变,甲基化结构的情况。
对于bulk RNA-seq,差异基因是同一基因在不同条件下(wild-type & mutant; stimulated & unstimulated)表达水平不同,(log2FC, fold-change )。
常用DEseq2,edgeR,limma包进行差异基因分析。
对于bulk RNA-seq分析出来的差异基因,可以通过实验进行验证,但是,scRNA-seq分析出来的差异基因,却缺乏验证。
对于scRNA-seq,不存在预先设定好的分组,因此需要通过聚类确定不同的细胞簇。通常会通过无监督方法聚类。
单细胞聚类形成不同的组,才能对比不同的组差异表达的基因。
目前对差异基因的分析主要存在两种方式:
- 通过预先确定的统计模型,或是拟合一个模型并在多组中进行比较。
- 利用非参数检验判断任何基因表达数据的情况。即通过非参数检验将观测到的表达值转成每个基因的排序。
通过预先的统计模型进行差异基因分析的方法有:
- Negative Binomial (负二项分布模型)
负二项分布拟合bulk RNA-seq data非常好,有文章表明,负二项分布拟合UMI标记的数据也不错。
但是,负二项分布不适用于全长转录组数据的分析,在这种该数据中,存在大量的零值数据。
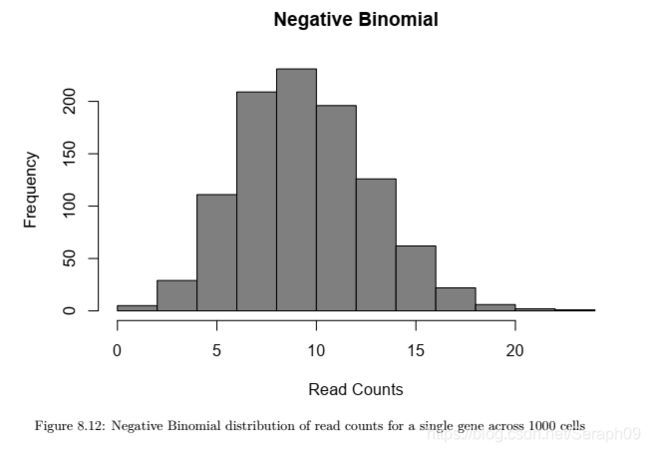
该图是负二项分布模型图
生成该模型的代码是:
set.seed(1)
hist(
rnbinom(
1000,
mu = 10,
size = 100),
col = "grey50",
xlab = "Read Counts",
main = "Negative Binomial"
)

2. 对于存在大量零值的数据,通常选用 zero-infalted negative binomial (零膨胀负二项分布),例如MAST,SCDE

生成该图的代码是
d <- 0.5;
counts <- rnbinom(
1000,
mu = 10,
size = 100
)
counts[runif(1000) < d] <- 0
hist(
counts,
col = "grey50",
xlab = "Read Counts",
main = "Zero-inflated NB"
)
![]()
![]()
d: the dropout rate
dropout rate和基因的平均表达水平相关。
- Possion-Beta分布
该方法基于transcriptional bursting, 这个模型比负二项分布的使用更不易。
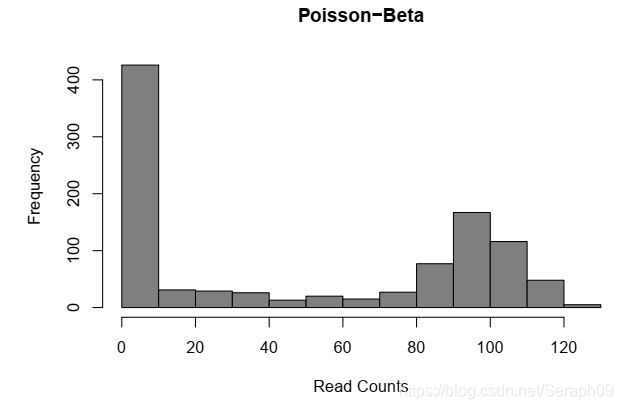
生成以上图形的代码是:
a <- 0.1
b <- 0.1
g <- 100
lambdas <- rbeta(1000, a, b)
counts <- sapply(g*lambdas, function(l) {rpois(1, lambda = l)})
hist(
counts,
col = "grey50",
xlab = "Read Counts",
main = "Poisson-Beta"
)
![]()
a: the rate of activation of transcription
b: the rate of inhibition of transcription
g: the rate of transcript production while transcription is active at the locus.
这些模型可能需要进一步发展用以解释基因表达中的batch-effect(批次效应),测序深度。
除却使用模型以外,可以采用非参数检验
5. Kolmogorov-Smirnov test
这是一种非参数检验,用于比较两个样本之间基因分布的差异。
-
Wilcox/Mann-Whitney-U Test
也是一种非参数检验,用于检验两组基因表达中位数之间的差异。和KS-test对于基因分布差异敏感不同。 -
edgeR
edgeR基于基因表达的负二项模型并使用广义线性模型框架。 -
Monocle
monocle使用多种不同的DE分析模型。
对于counts数据,则采用负二项分布模型。
对于标准化数据,进行对数变换后采用正态分布(高斯分布)。
该方法利用GLM模型,可用于解释batch effect. -
MAST
基于零膨胀负二项模型,并使用线性模型框架。
其它是运行速度比较慢(> 1h)的模型
-
BPSC:采用了泊松-beta模型,并与广义线性模型相结合。
-
SCDE:SCDE是第一个单细胞DE分析方法。它利用贝叶斯统计,采用了零膨胀负二项分布模型。
比较/结合scRNAseq 数据
寻找scRNA-seq数据
根据上述分析,我们逐渐设计一个理想的scRNA的流程
实验流程
处理reads序列
构建表达矩阵
生物学分析
Resources:
- scRNA-seq 方法
- ERCC
- scRNA-seq 分析工具
- scRNA-seq 的公共数据
练习数据:https://hemberg-lab.github.io/scRNA.seq.datasets/mouse/edev/#deng