因为课题就是做转录组测序的,所以基础知识有一些了解,接下来从数据处理部分开始进行笔记。
数据初步分析:
使用fastqc进行质量分析,这是一款Java软件,支持多线程。写这篇文章的时候版本是v0.11.7。
软件前期准备:
- 下载方式有两种:
官网下载好用filezilla导入linux服务器
直接在服务器中wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
- 接着安装
unzip fastqc_v0.11.7.zip-->cd FastQC-- >chmod755 fastqc
最后这个chmod有必要说一下,这个权限管理命令
chmod 用3个数字来表达对 用户(文件或目录的所有者),用户组(同组用户),其他用户 的权限:
如:chmod 755 fastqc
数字7是表达同时具有读,写,执行权限:(7 = 4 + 2+ 1)
读取--用数字4表示;
写入--用数字2表示;
执行--用数字1表示; 三者皆否:0
-
设置完权限后,还需要将FastQC文件夹(这里请注意是文件夹,而非fastqc这个可执行程序)导入环境变量
echo 'export PATH=/YOUR/FASTQC PATH/:$PATH' >> ~/.bashrc再
source ~/.bashrc 检查软件是否安装成功
fastqc --help出现帮助信息就可以使用啦!
后期软件使用:
基本格式 (各种参数+多个文件~支持多线程)
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] seqfile1 .. seqfileN
-o --outdir FastQC生成的报告文件的储存路径
--extract 使用这个参数是让程序不打包【默认会打包成一个压缩文件】
-t --threads 选择程序运行的线程数,每个线程会占用250MB内存(一般与文件数量一致就好)
-q --quiet 安静运行模式【不选这个选项,程序会实时报告运行的状况】
结果分析:
- 如果你有自己的转录组或者其他数据,可以现在测试了
- 如果没有,想学一下这个软件流程以及如何解读结果,可以下载公共数据(下载两个双端测序文件共4个)
# 顺便说一下这里用到了curl -O(保留远程文件的文件名) -L(对于自动跳转的网页,curl 就会跳转到新的网址)
# 当然用wget也可以,至于二者区别,可以参考https://www.cnblogs.com/lsdb/p/7171779.html
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1976948_1.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1976948_2.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1977249_1.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1977249_2.fastq.gz
# 下载完可以检查一下数据完整性,当然不是必须
md5sum *.gz
# 质控四个文件,我们可以采用四线程
# 大概用时 real 0m23.344s (如果你也想统计时间,就在命令前加time)
fastqc *.gz -t 4
#将结果.html用filezilla导出,浏览器查看
-
首先看到的是一个总结报告,
左边这一栏会告诉你,哪些是提出警告的(⚠️表示),那些是fail的(❌) image
image 接下来一部分一部分解析,共10部分
-
Basic Statistics 基本信息
 image
imageEncoding: 测序平台编号,现在Sanger/ Illumina 1.8以上都是Phred 33编码
Total sequences: reads数量(reads就是高通量测序平台产生的序列标签,翻译为读段!)
Sequence length: 测序长度
%GC: GC含量: 需要重点关注,可以帮助区别物种,人类细胞42%左右
Per base sequence quility:每个测序read各碱基质量【十分重要!】

横轴:测序序列的1-251个碱基;
纵轴:质量得分,score = -10 * log10(error),例如错误率error为1%,那么算出的score就是20
箱线图boxplot:对每一个碱基的质量的统计。箱子上面的须(up bar)为90%分位数,下面的须(down bar)为10%分位数,箱子中的红线为中位数即50%分位数,箱子顶(upside)为75%分位数,箱子低(downside)为25%分位数。这个boxplot的意义:一是看数据是否具有对称性;二是看数据分布差异,这里主要利用了第二点。bar的跨度越大,说明数据越不稳定。
蓝色的线将各个碱基的质量平均值连接起来
解释一下:图中蓝线的走势为何先高后低?因为目前采用的边合成边测序使用的是化学方法促使链由5'向3'延伸,也就是利用了DNA聚合酶。刚开始测序,合成反应还不是很稳定,但是酶的质量还很好,所以会在高质量区域内有一定的波动(这里的1-30bp),后来稳定了,但是随着时间的推移,酶的活力逐渐下降,特异性也变差,所以越往后出错几率越大。
【就像一个司机开车,一开始小心谨慎,起步慢,开的也慢,慢慢提速。后来越开越带劲,但是也越来越困,疲劳驾驶容易出事】一般能用的数据都要求至少Q20,也就是下四分位(10%分位数)的质量值要大于20。因此这里的189bp后面的需要切除,才能继续分析
二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%),Q30要求大于85%(最差也不要低于80%)
- Per sequence quility scores:每条序列 质量统计

横轴:质量值0-40,也即是Q值
纵轴:每个质量值对应的read数
我们的例子中一条read有251bp, 那么其中任意一条的251bp的质量平均值就是这条read的质量值。只要大部分都高于20说明比较正常
- Per base sequence content:read各个位置碱基比例分布

横轴:各碱基位置;纵轴:碱基百分比
四条线四种颜色代表四种碱基在每个位置的平均含量(一个位置会测很多reads,然后求一个平均)
一般来讲,A=T, C=G, 但是刚开始测序仪不稳定可能出现波动,这是正常的。一般不是波动特别大的,像这里cut掉前5bp就够了。另外如果A、T 或 C、G间出现偏差,只要在1%以内都是可以接受的
- Per sequence GC content: 序列平均GC分布
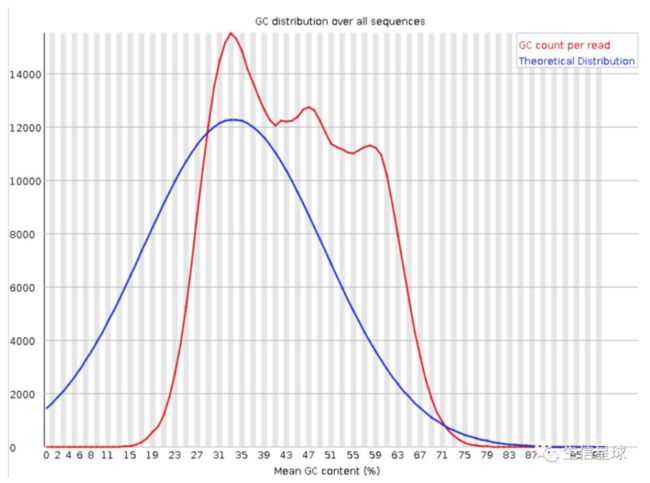
横轴为平均GC含量; 纵轴为每个GC含量对应的序列数量
蓝线为系统计算得到的理论分布;红线为测量值,二者越接近越好
-
这里不相符可能有两个原因:
前面提到了,GC可以作为物种特异性根据,这里出现了其他的峰有可能混入了其他物种的DNA;
目前二代测序基本都会有序列偏向性(所说的 bias),也就是某些特定区域会被反复测序,以至于高于正常水平,变相说明测序过程不够随机。这种现象会对以后的变异检测以及CNV分析造成影响
如果出现怎么办?-- 把和我们使用物种GC-content有差异的reads拿出来做blast,来确认是否为某些杂菌
- Per base N content: N含量分布

N是指仪器不能识别ATCG时给出的结果,一般不会出现。但是如果出现并且量还很大,应该就是测序系统或者试剂的问题
任意位置的N的比例超过5%,报"WARN";任意位置的N的比例超过20%,报"FAIL"
- Sequence length distribution: 序列长度统计

理想情况下,测得的序列长度应该是相等的。实际上总有些偏差
当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”
这里显示大部分都落在251bp这个测序长度上,有少量为250或252bp,但这不影响;如果偏差很大就不可信了
- Sequence duplication level:统计序列完全一样的reads的频率

横坐标是duplication的次数;纵坐标是duplicated reads的数目(红线)
解释下横坐标为何会有>10, >50等出现:测序的原始数据很大,如果每一条reads都统计,将耗时很久。这里软件只采用了数据的前200,000条reads统计其在全部数据中的重复数目,另外大于75bp的reads只取50bp进行比较。重复数大于10的reads被合并统计成了>10,以此类推...
unique reads总数(蓝线)作为100%,上图中可以看出,大概仅有2%的uniqe reads可以观察到两次重复。也就是说,我们这里的非unique reads占总数比例仅有2%左右。
-
正常情况下的确,测序深度越高,越容易产生一定程度的duplication。高程度的duplication level,提示我们可能有bias的存在(如建库过程中的PCR duplication)。
另外和做的项目也有关,一般转录组测序的结果中duplication level都比较高,60-70%都正常,这是因为转录组测的是基因的覆盖深度,各个基因表达量不同,如果某个基因覆盖度较高【tip:覆盖度是指基因/转录组测序测到的部分占整个组的比例】,那么测的部分就越多,相对应的duplication也会更高;对于外显子组测序来说,一般覆盖度比较一致,这里出现了duplication就不太正常。
当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“
- Overrepresented sequences:大量重复序列

和第8个duplication计算一样,也是取前200,000进行统计,大于75bp只取50bp。
发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“
- Adapter content: 接头含量

表示序列中两端adapter的情况
软件内置了四种常用的测序接头序列, fastqc 有一个参数-a可以自定义接头序列
此图中使用的illumina universal adapter并未去除,后期再使用trimmomatic/cutadapt去接头
我们在学习测序原理的时候知道了,接头的作用一个是能够使得序列结合到flowcell上,另外一个多样本测序时利用接头旁边的index加以区分
什么情况下能够测到接头呢? 一般测序read的长度大于插入片段(待测序列)的长度时会发生。对于WGS建库测序来讲,一般不会发生,因为他的待测序列要几百bp,而测序也就150bp算高了。但是对于RNA-seq,一般测序序列比较短,尤其是miRNA,只有几十bp,这是就会测到read末尾的接头
- (还有一类这里没体现)Kmer content: 重复短序列
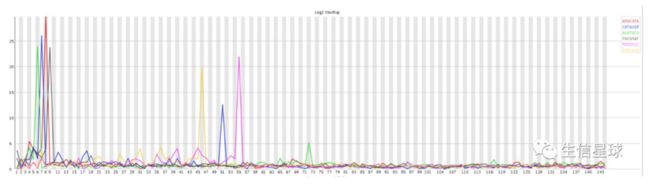
表示:在序列中某些特征的短序列重复出现的次数
-
这个图是转录组测序的一个文件,可以看到6-9bp几种短序列都出现了好多次。出现的原因可能是:
没有去除软件内置的adapter或者没有使用-a参数自定义adapter
序列本身重复度较高,例如在建库PCR过程出现序列偏向性bias--> 这在转录组测序中确实存在
数据过滤:
主要针对接头序列和低质量序列
工具: 有许多工具能干这事,例如SOAPnuke、cutadapt、untrimmed、sickle和seqtk等,其中经常用到的是Trimmomatic(也是一个java程序)
-
Trimmomatic:
-
安装:官网下载我使用的是0.36版本
 image
image下载后直接解压安装,其中有一个trimmomatic-0.36.jar是执行文件,使用时输入java -jar trimmomatic-0.36.jar就可以。另外还有一个adapter文件夹,里面存放了常用的illumina测序仪接头序列fasta格式,后续处理接头需要制定具体文件。
 image
image
-
-
关于adapter: 目前绝大部分的illumina的Hiseq和Miseq系列使用的都是Truseq3,过去的GA2测序仪使用的是Truseq2,PE/SE对应单端还是双端测序 , 如果使用的不是illumina测的,可以按照里面的格式自己新建一个接头文件,但其中的命名要注意。详情见官网主页
 image
image 以PE测序为例说一下命令参数设置:
java -jar PE [-threads ] log文件
双端测序原始fq文件
双端1输出文件 、 过滤掉的文件
双端2同理
<详细讲一下step:>
1\. ILLUMINACLIP: 根据上面qc部分的测试数据,我们设置TruSeq2-PE.fa:2:40:15
TruSeq2-PE.fa是接头序列;
2是比对时接头序列时所允许的最大错误数;
40是要求PE的两条read同时和adapter序列比对,匹配度加起来超40%,那么就认为这对PE reads含 有adapter,并在对应的位置需要进行切除;
【那么为何不是匹配100%?因为测序时并不是把 adapter全测了,只测了一部分】
15 指的是只要某条read的某部分与adapter超过了15%的相似度就认为包含,就要去除
2\. SLIDINGWINDOW: 滑动窗口长度 我们设置为4:20,代表窗口长度为4,窗口中的平均质量值至少为20,否 则会开始切除
3\. LEADING,指的是read开头的碱基是否要被切除的质量阈值,这里设为2
4\. TRAILING,指的是read末尾的碱基是否要被切除的质量阈值,这里设为2
5\. MINLEN,指的是read被切除后至少需要保留的长度,如果低于该长度,会被丢掉,这里设置25
#一个☝️范例(这个测试数据是phred 64的,所以使用默认值就好):
java -jar trimmomatic-0.36.jar PE -threads 8 \
-trimlog logfile \
reads_1.fq.gz reads_2.fq.gz \
out.read_1.fq.gz out.trim.read_1.fq.gz \
out.read_2.fq.gz out.trim.read_2.fq.gz \ ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq2-PE.fa:2:40:15 \
SLIDINGWINDOW:4:20 \
LEADING:2 TRAILING:2 \
MINLEN:25# 当然,这只是对一个样本的双端测序文件进行操作,那么问题来了:
# 如果你的样本比较多呢?还要手动一个个输入吗?
# 虽然说vim中使用快速替换 :%s/AA/BB/g 可以全局替换AA成BB,但还是有点麻烦
# 能不能让程序来一个自动化呢?是可以的!可以看作是上面脚本的改进版~
# 在脚本中构建for循环
# vi trim.sh
#开头这样写而不写*.gz的目的是避免了样本名称的重复
for filename in *_1.fastq.gz
do
#对于filename(%)向右匹配_的全部内容(*),然后这部分去掉,留下这之前的
base=${filename%_*}
java -jar trimmomatic-0.36.jar PE \ # 加上反斜线能让程序整体更清晰
-threads 8 \
-trimlog logfile \
${base}_1.fastq.gz ${base}_2.fastq.gz \
out.${base}_1.fastq.gz out.trim.${base}_1.fastq.gz \
out.${base}_2.fastq.gz out.trim.${base}_2.fastq.gz \
ILLUMINACLIP:/home/u1239/biosoft/Trimmomatic-0.36/adapters/TruSeq2-PE.fa:2:30:10 \
SLIDINGWINDOW:5:20 \
LEADING:5 TRAILING:5 \
MINLEN:50
done

-
如果报错,就说明参数错误例如我在运行的时候就有的问题百思不得解,敲了好几遍代码应该没错,但就是报错,换个服务器就好了,难道是平台问题?其实并不是,就是因为一点点小地方,有时只是一个代表换行的反斜线
\image
多样本质控:
fastqc对于一两个样品还能吃得消,但样本多了,我们如何同时查看对比他们的信息呢?
这里就可以使用界面更加优美的multiqc软件了, 他就是fastqc结果的整合我会从一个初学者角度来一步步进行,其中包括了中间犯的错误及改正~,如果你也在运行的过程发现了类似的错误,可以参考一下。
分界线1: 尝试自己安装
下载地址:
https://files.pythonhosted.org/packages/fa/3e/fbdcbaebadad2110eed3b4212ced58617cbd9badbfc728c5eabc53916b84/multiqc-1.5.tar.gz
解压缩后,会发现和以前安装的源代码文件不同,他没有直接显示可执行文件,也没有配置文件。这是因为multiqc是一个python软件,这里先看一下setup.py:

直接使用python setup.py会报错,因为可能你的服务器并没有setuptools,这是python依赖的第三方库。
先安装setuptools:
下载地址:
https://files.pythonhosted.org/packages/1a/04/d6f1159feaccdfc508517dba1929eb93a2854de729fa68da9d5c6b48fa00/setuptools-39.2.0.zip
解压缩完setuptools会看到有这些文件,这也是一般的python软件常见的:包括了setup.py,easy_install.py等

# 主要使用setup.py
python setup.py build # 编译
python setup.py install #安装
在第二步安装的时候报错,原因是不能对/usr/local/lib下的python进行操作,因为不管/usr/bin还是/usr/local/bin,都是可读不可改,如果自己家目录环境变量中查不到python的路径,那么安装过程中会自动调取更上层目录的python使用。这往往会引发一系列问题。如果要自己更改的话,需要自己home目录下有python
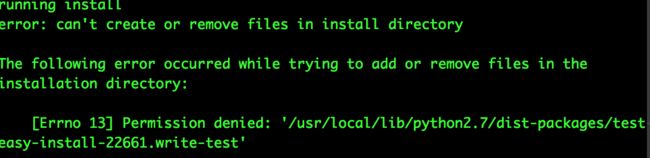
好吧,那么接下来我们再安装自己的python,毕竟自己的软件用起来方便:
# 下载地址
wget https://www.python.org/ftp/python/3.6.5/Python-3.6.5.tgz
# .tgz 就等同tar.gz
# 解压后会看到配置文件configure,设置成自己的软件环境变量即可
./configure --prefix=/YOUR PATH/
make
make install
# 可能结尾会有报错,但是不影响使用,出现了可执行文件python就成功了
# 将python复制到你的环境变量中就能直接调取
# 最后使用which python检测是否安在了自己的目录下
自己的python安装完成了,进入到解压好的multiqc文件夹下, python setup.py build 编译会运行成功,然后python setup.py install
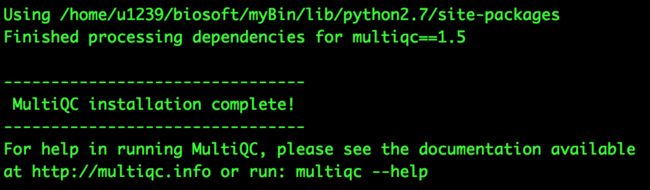
接着,multiqc就会出现在了自己的软件环境变量中了,输入multiqc就会看到

分界线2: 同一件事,换个玩法
目的:安装multiqc
途径:conda自动安装
缘由:好久不用conda安装软件,一直坚持源代码,因为好掌控。但是有的软件依赖的包很多,自己又很难发现这些潜在的关系,所以想重新试试conda,但并不是傻瓜式的使用,而是让conda听我的话~授之以鱼,不如授之以渔
Here we go!
安装: 安装过程需要注意
可以自定义新文件夹,默认是在home/miniconda3/
⚠️不要将conda添加到PATH中,我们只需要把它视作一个保姆babysitter,而不要当一个管家housekeeper
安装完conda,把miniconda/bin下的conda复制到你的/biotools/PATH(这个环境变量因人而异)
添加源: 清华源和中科大源都不错,别忘了再添加一个bioconda源
新建conda专属下载目录: 你可以在你的biotools目录下新建一个conda软件存放目录,例如conda_install。然后把这个文件夹添加到环境变量。以后你用conda安装的所有软件都存放在这里,
⚠️和你自己安装的软件要区分开, 然后利用conda install -p /PATH/conda_install multiqc 就可以实现multiqc的安装了
作者:刘小泽
链接:https://www.jianshu.com/p/101c14c3a1d2
来源:
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。