AlphaFold2.3 conda版本详细安装与使用
AlphaFold2.3 conda版本详细安装与使用
文章目录
- AlphaFold2.3 conda版本详细安装与使用
- 前言
- 一、安装环境
- 二、安装步骤
-
- 1. 确认已安装的cudatoolkit版本和cudnn版本
- 2. 创建虚拟环境alphafold-env并激活,指定python=3.8
- 3.安装AlphaFold2.3支持的新版openmm,CUDA等
- 4. clone alphafold并进入文件夹
- 5. 下载数据库及模型参数文件(参考官方说明)
- 6. 安装涉及到的依赖包。
- 7. 验证环境是否安装成功
- 8. 运行alphafold
- 9. 对alphafold预测结构使用Pymol根据pLDDT染色
- 安装-使用总结
前言
(1)结合AlphaFold官方提供了Docker安装指引,本文提供了conda版本的安装,配合run_alphafold.sh脚本使用更加方便。
(2)根据AlphaFold官方python脚本,更新了run_alphafold.sh中的models_to_relax参数,便于AF2.3版本使用。
(3)对alphafold预测结果使用Pymol根据pLDDT染色。
一、安装环境
本例安装环境:ubuntu 22.04, CUDA版本11.8,cudnn版本 8.6.0,RTX3060,4T机械硬盘。
二、安装步骤
1. 确认已安装的cudatoolkit版本和cudnn版本
通过nvidia-smi命令查看CUDA driver版本,如图为12.2。

通过 nvcc -V命令查看CUDA runtime版本,如图为11.8。
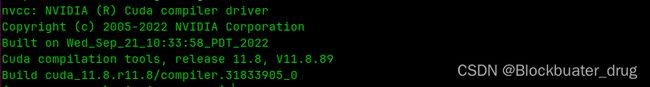
确保Alphafold2安装CUDA版本不高于CUDA runtime版本,即≤11.8。
通过 dpkg -l | grep cudnn 查看cudnn版本,本安装cudnn版本为8.6.0。
Note:确保CUDA和cudnn的版本匹配,TensorFlow版本需要与CUDA匹配。
2. 创建虚拟环境alphafold-env并激活,指定python=3.8
conda create -n alphafold-env python=3.8
conda activate alphafold-env
3.安装AlphaFold2.3支持的新版openmm,CUDA等
conda install -c conda-forge openmm==7.7.0 cudatoolkit==11.8.0
conda install -c bioconda hmmer==3.3.2 hhsuite==3.3.0 kalign2==2.04
conda install pdbfixer mock
4. clone alphafold并进入文件夹
git clone https://kkgithub.com/deepmind/alphafold.git
cd ./alphafold
#下载残基性质参数到common文件夹
wget -q -P alphafold/common/ https://git.scicore.unibas.ch/schwede/openstructure/-/raw/7102c63615b64735c4941278d92b554ec94415f8/modules/mol/alg/src/stereo_chemical_props.txt
5. 下载数据库及模型参数文件(参考官方说明)
#安装下载软件
sudo apt install aria2
使用官方脚本,位于alphafold/scripts/
sh scripts/download_all_data.sh <DOWNLOAD_DIR> > download.log 2> download_all.log &
NOTE: 保存数据库文件夹需要在alphafold主文件夹(即下文的 $data_dir)以外。
alphafold主文件夹可以位于任意位置,保持读写权限即可。
下载结束后,文件目录tree如下:

大小超过2.8T。
6. 安装涉及到的依赖包。
将如下内容保存到requirement.txt文件,使用pip安装。
absl-py==1.0.0
biopython==1.79
chex==0.0.7
dm-haiku==0.0.9
dm-tree==0.1.6
docker==5.0.0
immutabledict==2.0.0
ml-collections==0.1.0
pillow==10.1.0
numpy==1.21.6
pandas==1.3.4
scipy==1.7.3
tensorflow-gpu==2.11.0
zipp==3.17.0
jax==0.4.13
#根据安装情况调整
pip install -r requirement.txt
#安装jaxlib:与CUDA和cudnn硬件的版本可以查看https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
pip install --upgrade jaxlib==0.4.13+cuda11.cudnn86 -f https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
检查tensorflow-gpu安装:python
import tensorflow as tf
tf.config.list_physical_devices('GPU')
显示有类似如下信息,即为正常。
[PhysicalDevice(name='/physical_device:GPU:0', device_type='GPU')]
7. 验证环境是否安装成功
cd alphafold
#在主文件夹运行:
python run_alphafold_test.py
如有提示缺少安装文件,用conda或者pip安装。最终提示OK即可,示意如下图。

8. 运行alphafold
(1)使用run_alphafold.py运行:
对于单链蛋白(monomer)搜索full_dbs。
conda activate alphafold-env
cd alphafold
python run_alphafold.py \
--fasta_paths=/path/to/input/query.fasta \
--output_dir=/path/to/output/full \
--uniref90_database_path=$data_dir/uniref90/uniref90.fasta \
--mgnify_database_path=$data_dir/mgnify/mgy_clusters_2022_05.fa \
--data_dir=$data_dir \
--template_mmcif_dir=$data_dir/pdb_mmcif/mmcif_files \
--obsolete_pdbs_path=$data_dir/pdb_mmcif/obsolete.dat \
--pdb70_database_path=$data_dir/pdb70/pdb70 \
--uniref30_database_path=$data_dir/uniref30/UniRef30_2021_03 \
--bfd_database_path=$data_dir/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--max_template_date=2023-10-31 \
--models_to_relax=best \
--use_gpu_relax=true \
--db_preset=full_dbs \
--model_preset=monomer
(2)也可以使用run_alphafold.sh脚本便于输入,脚本从如下获取:
https://kkgithub.com/kalininalab/alphafold_non_docker/blob/main/run_alphafold.sh
对于AF2.1以后的版本,run_relax 已被models_to_relax替代,为了顺利运行run_alphafold.sh,笔者对 -r 参数做了修改,保持默认对最优输出结构(5个输出中pLDDT最高的1个)做relax(models_to_relax=“best”)。
新脚本命名为run_alphafold23.sh,脚本内容、使用方法和脚本使用示例如下。
#!/bin/bash
# Description: AlphaFold non-docker version
# Author: Sanjay Kumar Srikakulam
# modifier: Blockbuater_drug according to AF v2.3
usage() {
echo ""
echo "Please make sure all required parameters are given"
echo "Usage: $0 "
echo "Required Parameters:"
echo "-d Path to directory of supporting data"
echo "-o Path to a directory that will store the results."
echo "-f Path to FASTA files containing sequences. If a FASTA file contains multiple sequences, then it will be folded as a multimer. To fold more sequences one after another, write the files separated by a comma"
echo "-t Maximum template release date to consider (ISO-8601 format - i.e. YYYY-MM-DD). Important if folding historical test sets"
echo "Optional Parameters:"
echo "-g Enable NVIDIA runtime to run with GPUs (default: true)"
echo "-r For AF2.3 replacing run_relax by models_to_relax, " all", "none" and "best", "best" in default. Whether to run the final relaxation step on the predicted models. Turning relax off might result in predictions with distracting stereochemical violations but might help in case you are having issues with the relaxation stage (default: true)"
echo "-e Run relax on GPU if GPU is enabled (default: true)"
echo "-n OpenMM threads (default: all available cores)"
echo "-a Comma separated list of devices to pass to 'CUDA_VISIBLE_DEVICES' (default: 0)"
echo "-m Choose preset model configuration - the monomer model, the monomer model with extra ensembling, monomer model with pTM head, or multimer model (default: 'monomer')"
echo "-c Choose preset MSA database configuration - smaller genetic database config (reduced_dbs) or full genetic database config (full_dbs) (default: 'full_dbs')"
echo "-p Whether to read MSAs that have been written to disk. WARNING: This will not check if the sequence, database or configuration have changed (default: 'false')"
echo "-l How many predictions (each with a different random seed) will be generated per model. E.g. if this is 2 and there are 5 models then there will be 10 predictions per input. Note: this FLAG only applies if model_preset=multimer (default: 5)"
echo "-b Run multiple JAX model evaluations to obtain a timing that excludes the compilation time, which should be more indicative of the time required for inferencing many proteins (default: 'false')"
echo ""
exit 1
}
while getopts ":d:o:f:t:g:r:e:n:a:m:c:p:l:b:" i; do
case "${i}" in
d)
data_dir=$OPTARG
;;
o)
output_dir=$OPTARG
;;
f)
fasta_path=$OPTARG
;;
t)
max_template_date=$OPTARG
;;
g)
use_gpu=$OPTARG
;;
r)
models_to_relax=$OPTARG
;;
e)
enable_gpu_relax=$OPTARG
;;
n)
openmm_threads=$OPTARG
;;
a)
gpu_devices=$OPTARG
;;
m)
model_preset=$OPTARG
;;
c)
db_preset=$OPTARG
;;
p)
use_precomputed_msas=$OPTARG
;;
l)
num_multimer_predictions_per_model=$OPTARG
;;
b)
benchmark=$OPTARG
;;
esac
done
# Parse input and set defaults
if [[ "$data_dir" == "" || "$output_dir" == "" || "$fasta_path" == "" || "$max_template_date" == "" ]] ; then
usage
fi
if [[ "$benchmark" == "" ]] ; then
benchmark=false
fi
if [[ "$use_gpu" == "" ]] ; then
use_gpu=true
fi
if [[ "$gpu_devices" == "" ]] ; then
gpu_devices=0
fi
if [[ "$models_to_relax" == "" ]] ; then
models_to_relax="best"
fi
if [[ "$models_to_relax" != "best" && "$models_to_relax" != "all" && "$models_to_relax" != "none" ]] ; then
echo "Unknown models to relax preset! Using default ('best')"
db_preset="best"
fi
if [[ "$enable_gpu_relax" == "" ]] ; then
enable_gpu_relax="true"
fi
if [[ "$enable_gpu_relax" == true && "$use_gpu" == true ]] ; then
use_gpu_relax="true"
else
use_gpu_relax="false"
fi
if [[ "$num_multimer_predictions_per_model" == "" ]] ; then
num_multimer_predictions_per_model=5
fi
if [[ "$model_preset" == "" ]] ; then
model_preset="monomer"
fi
if [[ "$model_preset" != "monomer" && "$model_preset" != "monomer_casp14" && "$model_preset" != "monomer_ptm" && "$model_preset" != "multimer" ]] ; then
echo "Unknown model preset! Using default ('monomer')"
model_preset="monomer"
fi
if [[ "$db_preset" == "" ]] ; then
db_preset="full_dbs"
fi
if [[ "$db_preset" != "full_dbs" && "$db_preset" != "reduced_dbs" ]] ; then
echo "Unknown database preset! Using default ('full_dbs')"
db_preset="full_dbs"
fi
if [[ "$use_precomputed_msas" == "" ]] ; then
use_precomputed_msas="false"
fi
# This bash script looks for the run_alphafold.py script in its current working directory, if it does not exist then exits
current_working_dir=$(pwd)
alphafold_script="$current_working_dir/run_alphafold.py"
if [ ! -f "$alphafold_script" ]; then
echo "Alphafold python script $alphafold_script does not exist."
exit 1
fi
# Export ENVIRONMENT variables and set CUDA devices for use
# CUDA GPU control
export CUDA_VISIBLE_DEVICES=-1
if [[ "$use_gpu" == true ]] ; then
export CUDA_VISIBLE_DEVICES=0
if [[ "$gpu_devices" ]] ; then
export CUDA_VISIBLE_DEVICES=$gpu_devices
fi
fi
# OpenMM threads control
if [[ "$openmm_threads" ]] ; then
export OPENMM_CPU_THREADS=$openmm_threads
fi
# TensorFlow control
export TF_FORCE_UNIFIED_MEMORY='1'
# JAX control
export XLA_PYTHON_CLIENT_MEM_FRACTION='4.0'
# Path and user config (change me if required)
uniref90_database_path="$data_dir/uniref90/uniref90.fasta"
mgnify_database_path="$data_dir/mgnify/mgy_clusters_2022_05.fa"
template_mmcif_dir="$data_dir/pdb_mmcif/mmcif_files"
obsolete_pdbs_path="$data_dir/pdb_mmcif/obsolete.dat"
uniprot_database_path="$data_dir/uniprot/uniprot.fasta"
pdb_seqres_database_path="$data_dir/pdb_seqres/pdb_seqres.txt"
pdb70_database_path="$data_dir/pdb70/pdb70"
bfd_database_path="$data_dir/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt"
small_bfd_database_path="$data_dir/small_bfd/bfd-first_non_consensus_sequences.fasta"
uniref30_database_path="$data_dir/uniref30/UniRef30_2021_03"
# Binary path (change me if required)
hhblits_binary_path=$(which hhblits)
hhsearch_binary_path=$(which hhsearch)
jackhmmer_binary_path=$(which jackhmmer)
kalign_binary_path=$(which kalign)
binary_paths="--hhblits_binary_path=$hhblits_binary_path --hhsearch_binary_path=$hhsearch_binary_path --jackhmmer_binary_path=$jackhmmer_binary_path --kalign_binary_path=$kalign_binary_path"
database_paths="--uniref90_database_path=$uniref90_database_path --mgnify_database_path=$mgnify_database_path --data_dir=$data_dir --template_mmcif_dir=$template_mmcif_dir --obsolete_pdbs_path=$obsolete_pdbs_path"
if [[ $model_preset == "multimer" ]]; then
database_paths="$database_paths --uniprot_database_path=$uniprot_database_path --pdb_seqres_database_path=$pdb_seqres_database_path"
else
database_paths="$database_paths --pdb70_database_path=$pdb70_database_path"
fi
if [[ "$db_preset" == "reduced_dbs" ]]; then
database_paths="$database_paths --small_bfd_database_path=$small_bfd_database_path"
else
database_paths="$database_paths --uniref30_database_path=$uniref30_database_path --bfd_database_path=$bfd_database_path"
fi
command_args="--fasta_paths=$fasta_path --output_dir=$output_dir --max_template_date=$max_template_date --db_preset=$db_preset --model_preset=$model_preset --benchmark=$benchmark --use_precomputed_msas=$use_precomputed_msas --num_multimer_predictions_per_model=$num_multimer_predictions_per_model --models_to_relax=$models_to_relax --use_gpu_relax=$use_gpu_relax --logtostderr"
# Run AlphaFold with required parameters
$(python $alphafold_script $binary_paths $database_paths $command_args)
run_alphafold23.sh使用方法:
Usage: run_alphafold23.sh <OPTIONS>
Required Parameters:
-d <data_dir> Path to directory of supporting data
-o <output_dir> Path to a directory that will store the results.
-f <fasta_paths> Path to FASTA files containing sequences. If a FASTA file contains multiple sequences, then it will be folded as a multimer. To fold more sequences one after another, write the files separated by a comma
-t <max_template_date> Maximum template release date to consider (ISO-8601 format - i.e. YYYY-MM-DD). Important if folding historical test sets
Optional Parameters:
-g <use_gpu> Enable NVIDIA runtime to run with GPUs (default: true)
-r <models_to_relax> For AF2.3 replacing run_relax by models_to_relax, all, none and best, best in default. Whether to run the final relaxation step on the predicted models. Turning relax off might result in predictions with distracting stereochemical violations but might help in case you are having issues with the relaxation stage (default: true)
-e <enable_gpu_relax> Run relax on GPU if GPU is enabled (default: true)
-n <openmm_threads> OpenMM threads (default: all available cores)
-a <gpu_devices> Comma separated list of devices to pass to 'CUDA_VISIBLE_DEVICES' (default: 0)
-m <model_preset> Choose preset model configuration - the monomer model, the monomer model with extra ensembling, monomer model with pTM head, monomer_casp14 or multimer model (default: 'monomer')
-c <db_preset> Choose preset MSA database configuration - smaller genetic database config (reduced_dbs) or full genetic database config (full_dbs) (default: 'full_dbs')
-p <use_precomputed_msas> Whether to read MSAs that have been written to disk. WARNING: This will not check if the sequence, database or configuration have changed (default: 'false')
-l <num_multimer_predictions_per_model> How many predictions (each with a different random seed) will be generated per model. E.g. if this is 2 and there are 5 models then there will be 10 predictions per input. Note: this FLAG only applies if model_preset=multimer (default: 5)
-b <benchmark> Run multiple JAX model evaluations to obtain a timing that excludes the compilation time, which should be more indicative of the time required for inferencing many proteins (default: 'false')
run_alphafold23.sh使用示例:
将run_alphafold23.sh置于alphafold主目录。
下载rcsb_pdb_8I55.fasta文件(https://www.rcsb.org/fasta/entry/8I55)作为序列输入。
/path/to/inputdir和/path/to/outputdir分别为输入和输出文件夹路径。
conda activate alphafold-env
cd alphafold
bash ./run_alphafold23.sh -d $data_dir -o /path/to/outputdir -f /path/to/inputdir/rcsb_pdb_8I55.fasta -t 2023-10-31 -c reduced_dbs
运行~20min,结果文件夹中有以下输出文件:

ranking_debug.json :5种模型预测结构的pLDDT打分。
{
"plddts": {
"model_1_pred_0": 87.53338158106746,
"model_2_pred_0": 86.49349522466628,
"model_3_pred_0": 85.68943193089224,
"model_4_pred_0": 84.15560768736528,
"model_5_pred_0": 84.52065593375735
},
"order": [
"model_1_pred_0",
"model_2_pred_0",
"model_3_pred_0",
"model_5_pred_0",
"model_4_pred_0"
]
}
timings.json: 记录的运行时间如下,单位s。
{
"features": 1483.1720588207245,
"process_features_model_1_pred_0": 2.3148465156555176,
"predict_and_compile_model_1_pred_0": 85.52882599830627,
"process_features_model_2_pred_0": 1.143226146697998,
"predict_and_compile_model_2_pred_0": 80.66072988510132,
"process_features_model_3_pred_0": 1.0205607414245605,
"predict_and_compile_model_3_pred_0": 63.78041362762451,
"process_features_model_4_pred_0": 1.0224313735961914,
"predict_and_compile_model_4_pred_0": 63.5577335357666,
"process_features_model_5_pred_0": 1.0538318157196045,
"predict_and_compile_model_5_pred_0": 63.124531269073486,
"relax_model_1_pred_0": 5.963490009307861
}
confidence_model_1_pred_0.json :每个残基的打分。
{"residueNumber":[1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143],"confidenceScore":[93.52,97.19,98.5,98.72,98.51,98.37,97.37,95.5,91.07,88.46,88.55,89.87,91.54,91.29,90.86,89.49,94.56,96.18,94.89,96.9,97.53,97.97,97.87,98.19,98.32,98.35,97.94,97.83,98.04,97.64,96.6,97.23,97.59,97.23,98.33,98.62,98.37,98.3,96.92,95.2,91.22,87.58,88.01,87.34,91.07,95.15,94.87,95.57,95.05,96.04,97.44,97.35,96.61,97.26,98.01,97.39,96.96,97.79,97.62,98.04,97.87,98.31,98.5,98.54,98.3,97.84,97.28,96.61,94.93,94.55,93.86,91.7,89.53,85.18,85.64,86.04,87.81,89.16,90.58,92.12,92.65,91.3,90.9,92.83,90.74,89.27,87.36,75.99,64.56,57.37,51.47,39.82,47.69,40.84,47.46,35.5,47.18,44.85,41.38,48.78,42.01,51.79,49.66,51.94,51.03,48.19,51.79,52.1,57.6,64.85,71.12,84.97,88.06,92.0,94.13,93.41,95.49,97.16,97.29,97.29,97.52,97.3,97.12,95.08,95.7,95.48,93.76,94.57,95.46,97.16,96.88,96.5,96.89,97.32,96.75,97.27,98.05,97.98,96.94,97.82,97.79,94.74,87.27],"confidenceCategory":["H","H","H","H","H","H","H","H","H","M","M","M","H","H","H","M","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","M","M","M","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","M","M","M","M","M","M","H","H","H","H","H","H","H","M","M","M","L","L","L","D","D","D","D","D","D","D","D","D","D","L","D","L","L","D","L","L","L","L","M","M","M","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","H","M"]}
relaxed_model_1_pred_0.pdb:为relaxation之后的结构,相比增加了主链和侧链的氢原子,结构文件中第11列即为pLDDT,对应实验获得pdb文件中的b-factor一列。
relaxation阶段采用openmm amber_relaxer进行minimization,ff99sb力场,用到GPU。
9. 对alphafold预测结构使用Pymol根据pLDDT染色
Pymol安装可参考(ubuntu 安装 最新 PyMOL [源码安装][免费]):https://blog.csdn.net/zdx1996/article/details/121656769
打开Pymol,file→Edit pymolrc,确定,打开pymolrc,输入如下内容;点击file→Save,关闭.pymolrc窗口。
run https://raw.kkgithub.com/cbalbin-bio/pymol-color-alphafold/master/coloraf.py
在Pymol 中使用命令:coloraf relaxed_model_1_pred_0
结果如下图,蓝色blue和青色cyan标识的区域分别为预测残基置信度pLDDT>90和90>pLDDT>70,属于可信度高的区域。
 通过align,alphafold预测最优结果relaxation后,与X-ray pdb(8I55)结构中4个α-helix与4个β-sheet目视均预测良好,计算RMSD=0.35;
通过align,alphafold预测最优结果relaxation后,与X-ray pdb(8I55)结构中4个α-helix与4个β-sheet目视均预测良好,计算RMSD=0.35;
预测结构给出了loop可能的构象,相应这部分在X-ray结构中是缺失的,如下图。

安装-使用总结
(1)MSA阶段文件搜索较为耗时,硬盘读取速度较为重要;模型预测阶段minimization用到GPU,仅用CPU也可以,但耗时将显著增加;150AA蛋白Monomer搜索reduced_dbs,整个预测+relax在笔者硬件条件下大致需要25 min,同样条件full_dbs大致需要2 h。
(2)Multimer运行时间更久,30AA+150AA的双链Multimer,笔者硬件配置下预测时间超过了4.5 h;如要用来研究预测PPI,一定的硬件配置是必要的,尤其是硬盘和GPU。
以上就是AlphaFold2.3的conda环境安装及使用体验,欢迎批评指正与交流。