果子荐读
我们公众号集体休息了一个星期。下一次休息可能就是过年了。
昨天收到洲更的投稿,是因为上次我用Deseq2处理了一个TCGA的1222个样本。不同于 6个样本5分钟这种随心所欲的状态,这些样本处理了两次,最终还是挂在服务器上才完成了分析,前后花掉了几十个小时。然后,洲更跟我说,没必要。我说,Talk is cheap, show me your code。
这个,上门砸场子了,题目中的我是洲更,别人是我。
以下是正文
果子老师做过一个非常惊人的举动,用DESeq2处理1225例样本的TCGA数据,在没有使用DESeq多线程参数parallel的情况下,跑了将近40个小时。
那么问题来了,在那么大的样本量的情况下,应该用DESeq2进行数据处理吗?我的结论是不应该,DESeq2的适用场景是小样本的差异表分析,降低假阳性。当你的样本量足够多的时候,我们其实有更好的选择。
这里以果子老师的数据为例,来对比DESeq2的结果和我的分析结果进行比较.
先加载Dese2产生的dds, 这个dds的产生过程,可以参考这个帖子
TCGA提取非编码RNA并完成下游分析
load(file="dds_very_long.Rdata")
接下来用results函数提取差异分析结果,用table函数查看p值小于0.01分类结果
library(DESeq2)
deseq2_result <- results(dds)
## summary(deseq2_result, alpha = 0.01)
results <- data.frame(deseq2_result)
table(results$padj < 0.01)
加载表达量数据, 这个数据的名字叫expr_df
load(file = "BRCA_RNASEQ_exprdf.Rdata")

这是TCGA官网下载的数据读入R语言后的数据,可以通过这个帖子得到:
把GDC下载的多个TCGA文件批量读入R
用class看一下他的属性,知彼知己啊
class(expr_df)
[1] "data.table" "data.frame"
发现还有一个data.table属性,这个是因为我用fread的原因,因为他的语法跟data.frame有冲突,所以我们要把他转换一下
expr_df <- as.data.frame(expr_df)
把数据变成矩阵
expr_mt <- as.matrix(expr_df[,-1])
row.names(expr_mt) <- expr_df$gene_id
colnames(expr_mt) <- colnames(expr_df)[-1]
根据文库大小标准化(这一步需要拆解理解一下)
expr_mt <- expr_mt / rep(colSums(expr_mt), each=nrow(expr_mt)) * 1e6
过滤掉低表达量的基因
这里要求这个基因需要在超过1/3的样本中大于0(果子,这个条件是自己定的哈,我觉得在此刻有点苛刻了,如果正常组的样本没有那么多,而且有些基因本身就特异性表达,在这种严格的条件下,他们会被去掉。可以人为设置为2,意思是只要在两个样本中大于0即可)
index <- rowSums(expr_mt > 0) > (ncol(expr_mt) / 3)
table(index)
这样的基因还真不少
index
FALSE TRUE
25456 35027
把需要的样本提取出来
expr_mt <- expr_mt[index, ]
统计TCGA样本类型
TCGA_id <- colnames(expr_mt)
table(substring(TCGA_id,14,15))
01 06 11
1102 7 113
我们发现有7个样本是转移的,本次分析,我们关注的是癌症和癌旁,先把转移的样本去掉
TCGA_id <- TCGA_id[substring(TCGA_id,14,15)!="06"]
TCGA样本名称的第14,15代表样本的类型,具体的表格在这里
TGCA数据的标准化以及差异分析
创建分组信息
sample <- ifelse(substring(TCGA_id,14,15)=="01","cancer","normal")
sample <- factor(sample,levels = c("normal","cancer"),ordered = F)
metadata <- data.frame(TCGA_id,sample)
分别提取癌和癌旁的表达矩阵
cancer_sample <- metadata[metadata$sample == "cancer", "TCGA_id"]
normal_sample <- metadata[metadata$sample == "normal", "TCGA_id"]
cancer_df <- expr_mt[,colnames(expr_mt) %in% cancer_sample]
normal_df <- expr_mt[,colnames(expr_mt) %in% normal_sample]
取癌的表达量看一下,是这样的
计算logFC值
logFC <- log2(rowMeans(cancer_df) / rowMeans(normal_df))
下一步,利用非参数检验方法, wilcox.test,关于非参数检验的缘起可以看「女士品茶」的第16章摆脱参数
威尔科克森注释着计算t检验和方法分析的公式,意识到这些不同寻常的极端数值会对结果产生极大的影响,导致“学生”的t检验偏小。 ... 如果异常值体现了某种因素对系统数据的系统性污染,那么使用非参数方法只会让事情变得更糟。
主函数就是wilcox.test,他接受两组数据,可以返回p值
这里洲更用了apply+函数+并行化处理的方法
如果不会,就看看这个帖子及其中的链接:
迷人的多参数批量函数mapply
p_values <- future_lapply(seq(1,nrow(cancer_df)), function(x){
res <- wilcox.test(x = t(cancer_df[x,])[,1], y = t(normal_df[x,])[,1] )
res$p.value
})
用户 系统 流逝
3.30 3.87 9.75
最终用了10s不到就完成了哈
矫正p值
p <- unlist(p_values)
p.adj <- p.adjust(p, method = "fdr")
看一下结果
table(p.adj < 0.01)
FALSE TRUE
10997 24030
我们得到了24,030个校正后p值小于0.01的基因,而DESeq2是25,072个。如果比较全部的基因的话,韦恩图上可以发现,绝大部分基因都是相同的。

但是通常情况下,我们会更去关注一些变化比较大且p值显著的基因,用这些基因去做下游的富集分析。所以,下一步就是看看后面富集分析结果两者有什么区别。
我们用Y叔的clusterProfiler,去分析倍数变化4倍,矫正p值小于0.01的基因。
library(clusterProfiler)
library(org.Hs.eg.db)
org <- org.Hs.eg.db
diffgene1 <- row.names(expr_mt)[p.adj < 0.01 & abs(logFC) > 2]
diffgene1 <- substr(diffgene1, 1, 15)
diffgene2 <- row.names(deseq2_result)[deseq2_result$padj < 0.01 &
! is.na(deseq2_result$padj) &
abs(deseq2_result$log2FoldChange) > 2]
diffgene2 <- substr(diffgene2, 1, 15)
ego1 <- enrichGO(diffgene1,
OrgDb = org,
keyType = "ENSEMBL",
ont = "BP")
ego2 <- enrichGO(diffgene2,
OrgDb = org,
keyType = "ENSEMBL",
ont = "BP")
merge_result <- merge_result(list(wilcox=ego1,DESeq2=ego2))
dotplot(merge_result,showCategory= 20 )
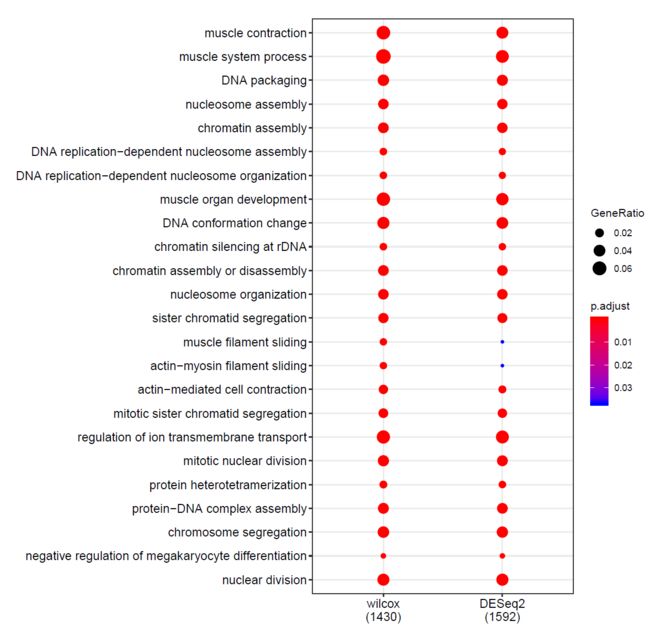
从点图中,你可以认为这两个分析结果是一致。
综上,当你在样本量足够多(两组都不少于30吧),其实没有去用DESeq2这些复杂的工具,用基础的统计学检验方法就能得到很好的结果了。
在样本量比较小的时候,用复杂的模型是无奈之举,它有很多假设成分在,尤其是你还想从无重复的实验设计中算p值。当你样本量够多的时候,用最简单的模型其实就会有很好的结果。
果子总结
这样看来,洲更说的是对的。不过,dds这里还要一个比较好的点就是,他可以做logFC矫正,就像这个帖子里面提到的,有一些count比较小,但是变化值很大的基因,会对GSEA分析产生影响
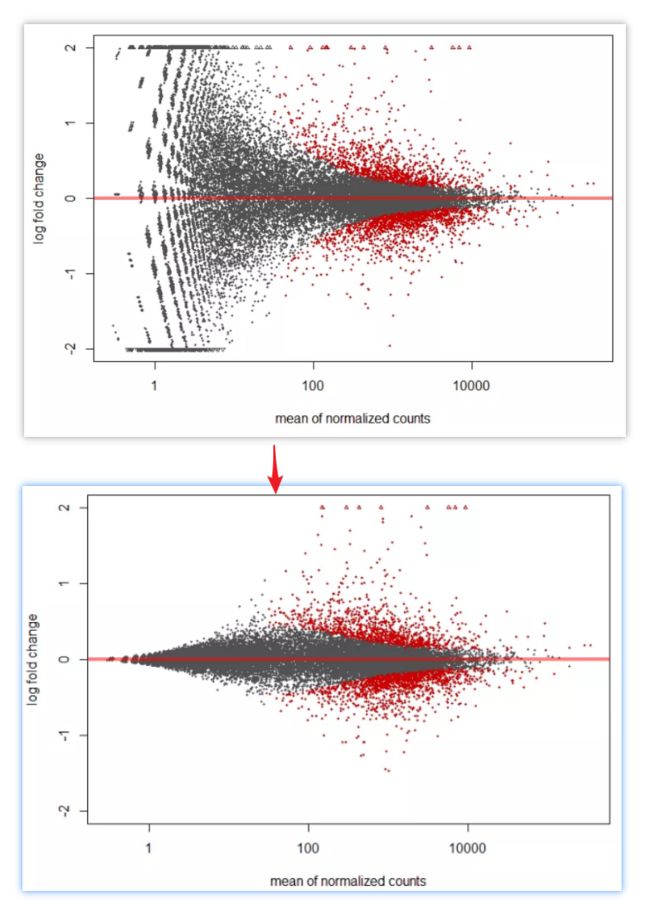
如果我们自己能够完成logFC矫正,那么,大样本的分析,就可以用这种简单的方法啦。
因为今天的帖子,很开放,所以,我们以R project形式提供了文中的原始数据,线下学员们肯定知道,提供R project文件,意味着数据,代码,结果全部在里面,大家可以方便地实现,并提出自己的想法(在公众号回复果子学统计,自助获取)
此外,9月广州,10月上海的线下课也来了。这次我们增加了不少新内容,期待见面。
久等了,果子学生信最新课程报名通知。