- 斤斤计较的婚姻到底有多难?
白心之岂必有为
很多人私聊我会问到在哪个人群当中斤斤计较的人最多?我都会回答他,一般婚姻出现问题的斤斤计较的人士会非常多,以我多年经验,在婚姻落的一塌糊涂的人当中,斤斤计较的人数占比在20~30%以上,也就是说10个婚姻出现问题的斤斤计较的人有2-3个有多不减。在婚姻出问题当中,有大量的心理不平衡的、尖酸刻薄的怨妇。在婚姻中仅斤斤计较有两种类型:第一种是物质上的,另一种是精神上的。在物质与精神上抠门已经严重的影响
- 情绪觉察日记第37天
露露_e800
今天是家庭关系规划师的第二阶最后一天,慧萍老师帮我做了个案,帮我处理了埋在心底好多年的一份恐惧,并给了我深深的力量!这几天出来学习,爸妈过来婆家帮我带小孩,妈妈出于爱帮我收拾东西,并跟我先生和婆婆产生矛盾,妈妈觉得他们没有照顾好我…。今晚回家见到妈妈,我很欣赏她并赞扬她,妈妈说今晚要跟我睡我说好,当我们俩躺在床上准备睡觉的时候,我握着妈妈的手对她说:妈妈这几天辛苦你了,你看你多利害把我们的家收拾得
- QQ群采集助手,精准引流必备神器
2401_87347160
其他经验分享
功能概述微信群查找与筛选工具是一款专为微信用户设计的辅助工具,它通过关键词搜索功能,帮助用户快速找到相关的微信群,并提供筛选是否需要验证的群组的功能。主要功能关键词搜索:用户可以输入关键词,工具将自动查找包含该关键词的微信群。筛选功能:工具提供筛选机制,用户可以选择是否只显示需要验证或不需要验证的群组。精准引流:通过上述功能,用户可以更精准地找到目标群组,进行有效的引流操作。3.设备需求该工具可以
- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- 铭刻于星(四十二)
随风至
69夜晚,绍敏同学做完功课后,看了眼房外,没听到动静才敢从书包的夹层里拿出那个心形纸团。折痕压得很深,都有些旧了,想来是已经写好很久了。绍敏同学慢慢地、轻轻地捏开折叠处,待到全部拆开后,又反复抚平纸张,然后仔细地一字字默看。只是开头的三个字是第一次看到,让她心漏跳了几拍。“亲爱的绍敏:从四年级的时候,我就喜欢你了,但是我一直不敢说,怕影响你学习。六年级的时候听说有人跟你表白,你接受了,我很难过,但
- 随笔 | 仙一般的灵气
海思沧海
仙岛今天,我看了你全部,似乎已经进入你的世界我不知道,这是否是梦幻,还是你仙一般的灵气吸引了我也许每一个人都要有一份属于自己的追求,这样才能够符合人生的梦想,生活才能够充满着阳光与快乐我不知道,我为什么会这样的感叹,是在感叹自己的人生,还是感叹自己一直没有孜孜不倦的追求只感觉虚度了光阴,每天活在自己的梦中,活在一个不真实的世界是在逃避自己,还是在逃避周围的一切有时候我嘲笑自己,嘲笑自己如此的虚无,
- 一百九十四章. 自相矛盾
巨木擎天
唉!就这么一夜,林子感觉就像过了很多天似的,先是回了阳间家里,遇到了那么多不可思议的事情儿。特别是小伙伴们,第二次与自己见面时,僵硬的表情和恐怖的气氛,让自己如坐针毡,打从心眼里难受!还有东子,他现在还好吗?有没有被人欺负?护城河里的小鱼小虾们,还都在吗?水不会真的干枯了吧?那对相亲相爱漂亮的太平鸟儿,还好吧!春天了,到了做窝、下蛋、喂养小鸟宝宝的时候了,希望它们都能够平安啊!虽然没有看见家人,也
- UI学习——cell的复用和自定义cell
Magnetic_h
ui学习
目录cell的复用手动(非注册)自动(注册)自定义cellcell的复用在iOS开发中,单元格复用是一种提高表格(UITableView)和集合视图(UICollectionView)滚动性能的技术。当一个UITableViewCell或UICollectionViewCell首次需要显示时,如果没有可复用的单元格,则视图会创建一个新的单元格。一旦这个单元格滚动出屏幕,它就不会被销毁。相反,它被添
- 微服务下功能权限与数据权限的设计与实现
nbsaas-boot
微服务java架构
在微服务架构下,系统的功能权限和数据权限控制显得尤为重要。随着系统规模的扩大和微服务数量的增加,如何保证不同用户和服务之间的访问权限准确、细粒度地控制,成为设计安全策略的关键。本文将讨论如何在微服务体系中设计和实现功能权限与数据权限控制。1.功能权限与数据权限的定义功能权限:指用户或系统角色对特定功能的访问权限。通常是某个用户角色能否执行某个操作,比如查看订单、创建订单、修改用户资料等。数据权限:
- 学点心理知识,呵护孩子健康
静候花开_7090
昨天听了华中师范大学教育管理学系副教授张玲老师的《哪里才是学生心理健康的最后庇护所,超越教育与技术的思考》的讲座。今天又重新学习了一遍,收获匪浅。张玲博士也注意到了当今社会上的孩子由于心理问题导致的自残、自杀及伤害他人等恶性事件。她向我们普及了一个重要的命题,她说心理健康的一些基本命题,我们与我们通常的一些教育命题是不同的,她还举了几个例子,让我们明白我们原来以为的健康并非心理学上的健康。比如如果
- 《投行人生》读书笔记
小蘑菇的树洞
《投行人生》----作者詹姆斯-A-朗德摩根斯坦利副主席40年的职业洞见-很短小精悍的篇幅,比较适合初入职场的新人。第一部分成功的职业生涯需要规划1.情商归为适应能力分享与协作同理心适应能力,更多的是自我意识,你有能力识别自己的情并分辨这些情绪如何影响你的思想和行为。2.对于初入职场的人的建议,细节,截止日期和数据很重要截止日期,一种有效的方法是请老板为你所有的任务进行优先级排序。和老板喝咖啡的好
- 《策划经理回忆录之二》
路基雅虎
话说三年变六年,飘了,飘了……眨眼,2013年5月,老吴回到了他的家乡——油城从新开启他的工作幻想症生涯。很庆幸,这是一家很有追求,同时敢于尝试的,且实力不容低调的新星房企——金源置业(前身泰源置业)更值得庆幸的是第一个盘就是油城十路的标杆之一:金源盛世。2013年5月,到2015年11月,两年的陪伴,迎来了一场大爆发。2000个筹,5万/筹,直接回笼1个亿!!!这……让我开始认真审视这座看似五线
- Long类型前后端数据不一致
igotyback
前端
响应给前端的数据浏览器控制台中response中看到的Long类型的数据是正常的到前端数据不一致前后端数据类型不匹配是一个常见问题,尤其是当后端使用Java的Long类型(64位)与前端JavaScript的Number类型(最大安全整数为2^53-1,即16位)进行数据交互时,很容易出现精度丢失的问题。这是因为JavaScript中的Number类型无法安全地表示超过16位的整数。为了解决这个问
- 店群合一模式下的社区团购新发展——结合链动 2+1 模式、AI 智能名片与 S2B2C 商城小程序源码
说私域
人工智能小程序
摘要:本文探讨了店群合一的社区团购平台在当今商业环境中的重要性和优势。通过分析店群合一模式如何将互联网社群与线下终端紧密结合,阐述了链动2+1模式、AI智能名片和S2B2C商城小程序源码在这一模式中的应用价值。这些创新元素的结合为社区团购带来了新的机遇,提升了用户信任感、拓展了营销渠道,并实现了线上线下的完美融合。一、引言随着互联网技术的不断发展,社区团购作为一种新兴的商业模式,在满足消费者日常需
- 2021-08-26
影幽
在生活中,女人与男人的感悟往往有所不同。人生最大的舞台就是生活,大幕随时都可能拉开,关键是你愿不愿意表演都无法躲避。在生活中,遇事不要急躁,不要急于下结论,尤其生气时不要做决断,要学会换位思考,大事化小小事化了,把复杂的事情尽量简单处理,千万不要把简单的事情复杂化。永远不要扭曲,别人善意,无药可救。昨天是张过期的支票,明天是张信用卡,只有今天才是现金,要善加利用!执着的攀登者不必去与别人比较自己的
- ArcGIS栅格计算器常见公式(赋值、0和空值的转换、补充栅格空值)
研学随笔
arcgis经验分享
我们在使用ArcGIS时通常经常用到栅格计算器,今天主要给大家介绍我日常中经常用到的几个公式,供大家参考学习。将特定值(-9999)赋值为0,例如-9999.Con("raster"==-9999,0,"raster")2.给空值赋予特定的值(如0)Con(IsNull("raster"),0,"raster")3.将特定的栅格值(如1)赋值为空值,其他保留原值SetNull("raster"==
- 高级编程--XML+socket练习题
masa010
java开发语言
1.北京华北2114.8万人上海华东2,500万人广州华南1292.68万人成都华西1417万人(1)使用dom4j将信息存入xml中(2)读取信息,并打印控制台(3)添加一个city节点与子节点(4)使用socketTCP协议编写服务端与客户端,客户端输入城市ID,服务器响应相应城市信息(5)使用socketTCP协议编写服务端与客户端,客户端要求用户输入city对象,服务端接收并使用dom4j
- 2018-07-23-催眠日作业-#不一样的31天#-66小鹿
小鹿_33
预言日:人总是在逃避命运的路上,与之不期而遇。心理学上有个著名的名词,叫做自证预言;经济学上也有一个很著名的定律叫做,墨菲定律;在灵修派上,还有一个很著名的法则,叫做吸引力法则。这3个领域的词,虽然看起来不太一样,但是他们都在告诉人们一个现象:你越担心什么,就越有可能会发生什么。同样的道理,你越想得到什么,就应该要积极地去创造什么。无论是自证预言,墨菲定律还是吸引力法则,对人都有正反2个维度的影响
- 【一起学Rust | 设计模式】习惯语法——使用借用类型作为参数、格式化拼接字符串、构造函数
广龙宇
一起学Rust#Rust设计模式rust设计模式开发语言
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档文章目录前言一、使用借用类型作为参数二、格式化拼接字符串三、使用构造函数总结前言Rust不是传统的面向对象编程语言,它的所有特性,使其独一无二。因此,学习特定于Rust的设计模式是必要的。本系列文章为作者学习《Rust设计模式》的学习笔记以及自己的见解。因此,本系列文章的结构也与此书的结构相同(后续可能会调成结构),基本上分为三个部分
- 放下是一段成长的修行
小莳玥
人来到这个世界上,只有两件事:生和死。一件事已经做完了,另一件你还急什么呢?是人,都有七情六欲。是心,都有喜怒哀乐,这些再正常不过了。别总抱怨自己活得累,过得辛苦。永远记住:舒坦是留给死人的。苦,才是生活;累,才是工作;变,才是命运;忍,才是历练;容,才是智慧;静,才是修养;舍,才会得到;做,才会拥有。人生,活得太清楚,才是最大的不明白。有些事,看得很清,却说不清;有些人,了解很深,却猜不透;有些
- 回溯 Leetcode 332 重新安排行程
mmaerd
Leetcode刷题学习记录leetcode算法职场和发展
重新安排行程Leetcode332学习记录自代码随想录给你一份航线列表tickets,其中tickets[i]=[fromi,toi]表示飞机出发和降落的机场地点。请你对该行程进行重新规划排序。所有这些机票都属于一个从JFK(肯尼迪国际机场)出发的先生,所以该行程必须从JFK开始。如果存在多种有效的行程,请你按字典排序返回最小的行程组合。例如,行程[“JFK”,“LGA”]与[“JFK”,“LGB
- 每日一题——第九十题
互联网打工人no1
C语言程序设计每日一练c语言
题目:判断子串是否与主串匹配#include#include#include//////判断子串是否在主串中匹配//////主串///子串///boolisSubstring(constchar*str,constchar*substr){intlenstr=strlen(str);//计算主串的长度intlenSub=strlen(substr);//计算子串的长度//遍历主字符串,对每个可能得
- Python数据分析与可视化实战指南
William数据分析
pythonpython数据
在数据驱动的时代,Python因其简洁的语法、强大的库生态系统以及活跃的社区,成为了数据分析与可视化的首选语言。本文将通过一个详细的案例,带领大家学习如何使用Python进行数据分析,并通过可视化来直观呈现分析结果。一、环境准备1.1安装必要库在开始数据分析和可视化之前,我们需要安装一些常用的库。主要包括pandas、numpy、matplotlib和seaborn等。这些库分别用于数据处理、数学
- 《庄子.达生9》
钱江潮369
【原文】孔子观于吕梁,县水三十仞,流沫四十里,鼋鼍鱼鳖之所不能游也。见一丈夫游之,以为有苦而欲死也,使弟子并流而拯之。数百步而出,被发行歌而游于塘下。孔子从而问焉,曰:“吾以子为鬼,察子则人也。请问,‘蹈水有道乎’”曰:“亡,吾无道。吾始乎故,长乎性,成乎命。与齐俱入,与汩偕出,从水之道而不为私焉。此吾所以蹈之也。”孔子曰:“何谓始乎故,长乎性,成乎命?”曰:“吾生于陵而安于陵,故也;长于水而安于
- 水泥质量纠纷案代理词
徐宝峰律师
贵州领航建设有限公司诉贵州纳雍隆庆乌江水泥有限公司产品质量纠纷案代理词尊敬的审判长、审判员:贵州千里律师事务所接受被告贵州纳雍隆庆乌江水泥有限公司的委托,指派我担任其诉讼代理人,参加本案的诉讼活动。下面,我结合本案事实和相关法律规定发表如下代理意见,供合议庭评议案件时参考:原告应当举证证明其遭受的损失与被告生产的水泥质量的因果关系。首先水泥是一种粉状水硬性无机胶凝材料。加水搅拌后成浆体,能在空气中
- 2019-12-22-22:30
涓涓1016
今天是冬至,写下我的日更,是因为这两天的学习真的是能量的满满,让我看到了自己,未来另外一种可能性,也让我看到了这两年这几年的过程中我所接受那些痛苦的来源。一切的根源和痛苦都来自于人生,家庭,而你的原生家庭,你的爸爸和妈妈,是因为你这个灵魂在那一刻选择他们作为你的爸爸和妈妈来的,所以你得接受他,你得接纳他,他就是因为他的存在而给你的学习和成长带来这些痛苦,那其实是你必然要经历的这个过程,当你去接纳的
- Goolge earth studio 进阶4——路径修改与平滑
陟彼高冈yu
Googleearthstudio进阶教程旅游
如果我们希望在大约中途时获得更多的城市鸟瞰视角。可以将相机拖动到这里并创建一个新的关键帧。camera_target_clip_7EarthStudio会自动平滑我们的路径,所以当我们通过这个关键帧时,不是一个生硬的角度,而是一个平滑的曲线。camera_target_clip_8路径上有贝塞尔控制手柄,允许我们调整路径的形状。右键单击,我们可以选择“平滑路径”,这是默认的自动平滑算法,或者我们可
- 将cmd中命令输出保存为txt文本文件
落难Coder
Windowscmdwindow
最近深度学习本地的训练中我们常常要在命令行中运行自己的代码,无可厚非,我们有必要保存我们的炼丹结果,但是复制命令行输出到txt是非常麻烦的,其实Windows下的命令行为我们提供了相应的操作。其基本的调用格式就是:运行指令>输出到的文件名称或者具体保存路径测试下,我打开cmd并且ping一下百度:pingwww.baidu.com>./data.txt看下相同目录下data.txt的输出:如果你再
- 18-115 一切思考不能有效转化为行动,都TM是扯淡!
成长时间线
7月25号写了一篇关于为什么会断更如此严重的反思,然而,之后日更仅仅维持了一周,又出现了这次更严重的现象。从8月2号到昨天8月6号,5天!又是5天没有更文!虽然这次断更时间和上次一样,那为什么说这次更严重?因为上次之后就分析了问题的原因,以及应该如何解决,按理说应该会好转,然而,没过几天严重断更的现象再次出现,想想,经过反思,问题依然没有解决与改变,这让我有些担忧。到底是哪里出了问题,难道我就真的
- 山东大学小树林支教调研团青青仓木队——翟晓楠
山东大学青青仓木队
过了半年,又一次启程,又一次回到支教的初心之地。比起上一次的试探与不安,我更多了一丝稳重与熟练。心境、处境也都随着半个学期的过去而变得不同,半个学期中,身体上的,心理上的,太多的逆境让我变得步履维艰,曲曲折折,弯弯绕绕,我仿佛打不起精神,没有胃口,没有动力。感觉走的不顺畅的时候,支教这个旅程,给了我力量。自告奋勇承担起队长这一职务的我,从组织时的复杂和困难的经历,协调各种问题,从无到有,和校长和队
- 算法 单链的创建与删除
换个号韩国红果果
c算法
先创建结构体
struct student {
int data;
//int tag;//标记这是第几个
struct student *next;
};
// addone 用于将一个数插入已从小到大排好序的链中
struct student *addone(struct student *h,int x){
if(h==NULL) //??????
- 《大型网站系统与Java中间件实践》第2章读后感
白糖_
java中间件
断断续续花了两天时间试读了《大型网站系统与Java中间件实践》的第2章,这章总述了从一个小型单机构建的网站发展到大型网站的演化过程---整个过程会遇到很多困难,但每一个屏障都会有解决方案,最终就是依靠这些个解决方案汇聚到一起组成了一个健壮稳定高效的大型系统。
看完整章内容,
- zeus持久层spring事务单元测试
deng520159
javaDAOspringjdbc
今天把zeus事务单元测试放出来,让大家指出他的毛病,
1.ZeusTransactionTest.java 单元测试
package com.dengliang.zeus.webdemo.test;
import java.util.ArrayList;
import java.util.List;
import org.junit.Test;
import
- Rss 订阅 开发
周凡杨
htmlxml订阅rss规范
RSS是 Really Simple Syndication的缩写(对rss2.0而言,是这三个词的缩写,对rss1.0而言则是RDF Site Summary的缩写,1.0与2.0走的是两个体系)。
RSS
- 分页查询实现
g21121
分页查询
在查询列表时我们常常会用到分页,分页的好处就是减少数据交换,每次查询一定数量减少数据库压力等等。
按实现形式分前台分页和服务器分页:
前台分页就是一次查询出所有记录,在页面中用js进行虚拟分页,这种形式在数据量较小时优势比较明显,一次加载就不必再访问服务器了,但当数据量较大时会对页面造成压力,传输速度也会大幅下降。
服务器分页就是每次请求相同数量记录,按一定规则排序,每次取一定序号直接的数据
- spring jms异步消息处理
510888780
jms
spring JMS对于异步消息处理基本上只需配置下就能进行高效的处理。其核心就是消息侦听器容器,常用的类就是DefaultMessageListenerContainer。该容器可配置侦听器的并发数量,以及配合MessageListenerAdapter使用消息驱动POJO进行消息处理。且消息驱动POJO是放入TaskExecutor中进行处理,进一步提高性能,减少侦听器的阻塞。具体配置如下:
- highCharts柱状图
布衣凌宇
hightCharts柱图
第一步:导入 exporting.js,grid.js,highcharts.js;第二步:写controller
@Controller@RequestMapping(value="${adminPath}/statistick")public class StatistickController { private UserServi
- 我的spring学习笔记2-IoC(反向控制 依赖注入)
aijuans
springmvcSpring 教程spring3 教程Spring 入门
IoC(反向控制 依赖注入)这是Spring提出来了,这也是Spring一大特色。这里我不用多说,我们看Spring教程就可以了解。当然我们不用Spring也可以用IoC,下面我将介绍不用Spring的IoC。
IoC不是框架,她是java的技术,如今大多数轻量级的容器都会用到IoC技术。这里我就用一个例子来说明:
如:程序中有 Mysql.calss 、Oracle.class 、SqlSe
- TLS java简单实现
antlove
javasslkeystoretlssecure
1. SSLServer.java
package ssl;
import java.io.FileInputStream;
import java.io.InputStream;
import java.net.ServerSocket;
import java.net.Socket;
import java.security.KeyStore;
import
- Zip解压压缩文件
百合不是茶
Zip格式解压Zip流的使用文件解压
ZIP文件的解压缩实质上就是从输入流中读取数据。Java.util.zip包提供了类ZipInputStream来读取ZIP文件,下面的代码段创建了一个输入流来读取ZIP格式的文件;
ZipInputStream in = new ZipInputStream(new FileInputStream(zipFileName));
&n
- underscore.js 学习(一)
bijian1013
JavaScriptunderscore
工作中需要用到underscore.js,发现这是一个包括了很多基本功能函数的js库,里面有很多实用的函数。而且它没有扩展 javascript的原生对象。主要涉及对Collection、Object、Array、Function的操作。 学
- java jvm常用命令工具——jstatd命令(Java Statistics Monitoring Daemon)
bijian1013
javajvmjstatd
1.介绍
jstatd是一个基于RMI(Remove Method Invocation)的服务程序,它用于监控基于HotSpot的JVM中资源的创建及销毁,并且提供了一个远程接口允许远程的监控工具连接到本地的JVM执行命令。
jstatd是基于RMI的,所以在运行jstatd的服务
- 【Spring框架三】Spring常用注解之Transactional
bit1129
transactional
Spring可以通过注解@Transactional来为业务逻辑层的方法(调用DAO完成持久化动作)添加事务能力,如下是@Transactional注解的定义:
/*
* Copyright 2002-2010 the original author or authors.
*
* Licensed under the Apache License, Version
- 我(程序员)的前进方向
bitray
程序员
作为一个普通的程序员,我一直游走在java语言中,java也确实让我有了很多的体会.不过随着学习的深入,java语言的新技术产生的越来越多,从最初期的javase,我逐渐开始转变到ssh,ssi,这种主流的码农,.过了几天为了解决新问题,webservice的大旗也被我祭出来了,又过了些日子jms架构的activemq也开始必须学习了.再后来开始了一系列技术学习,osgi,restful.....
- nginx lua开发经验总结
ronin47
使用nginx lua已经两三个月了,项目接开发完毕了,这几天准备上线并且跟高德地图对接。回顾下来lua在项目中占得必中还是比较大的,跟PHP的占比差不多持平了,因此在开发中遇到一些问题备忘一下 1:content_by_lua中代码容量有限制,一般不要写太多代码,正常编写代码一般在100行左右(具体容量没有细心测哈哈,在4kb左右),如果超出了则重启nginx的时候会报 too long pa
- java-66-用递归颠倒一个栈。例如输入栈{1,2,3,4,5},1在栈顶。颠倒之后的栈为{5,4,3,2,1},5处在栈顶
bylijinnan
java
import java.util.Stack;
public class ReverseStackRecursive {
/**
* Q 66.颠倒栈。
* 题目:用递归颠倒一个栈。例如输入栈{1,2,3,4,5},1在栈顶。
* 颠倒之后的栈为{5,4,3,2,1},5处在栈顶。
*1. Pop the top element
*2. Revers
- 正确理解Linux内存占用过高的问题
cfyme
linux
Linux开机后,使用top命令查看,4G物理内存发现已使用的多大3.2G,占用率高达80%以上:
Mem: 3889836k total, 3341868k used, 547968k free, 286044k buffers
Swap: 6127608k total,&nb
- [JWFD开源工作流]当前流程引擎设计的一个急需解决的问题
comsci
工作流
当我们的流程引擎进入IRC阶段的时候,当循环反馈模型出现之后,每次循环都会导致一大堆节点内存数据残留在系统内存中,循环的次数越多,这些残留数据将导致系统内存溢出,并使得引擎崩溃。。。。。。
而解决办法就是利用汇编语言或者其它系统编程语言,在引擎运行时,把这些残留数据清除掉。
- 自定义类的equals函数
dai_lm
equals
仅作笔记使用
public class VectorQueue {
private final Vector<VectorItem> queue;
private class VectorItem {
private final Object item;
private final int quantity;
public VectorI
- Linux下安装R语言
datageek
R语言 linux
命令如下:sudo gedit /etc/apt/sources.list1、deb http://mirrors.ustc.edu.cn/CRAN/bin/linux/ubuntu/ precise/ 2、deb http://dk.archive.ubuntu.com/ubuntu hardy universesudo apt-key adv --keyserver ke
- 如何修改mysql 并发数(连接数)最大值
dcj3sjt126com
mysql
MySQL的连接数最大值跟MySQL没关系,主要看系统和业务逻辑了
方法一:进入MYSQL安装目录 打开MYSQL配置文件 my.ini 或 my.cnf查找 max_connections=100 修改为 max_connections=1000 服务里重起MYSQL即可
方法二:MySQL的最大连接数默认是100客户端登录:mysql -uusername -ppass
- 单一功能原则
dcj3sjt126com
面向对象的程序设计软件设计编程原则
单一功能原则[
编辑]
SOLID 原则
单一功能原则
开闭原则
Liskov代换原则
接口隔离原则
依赖反转原则
查
论
编
在面向对象编程领域中,单一功能原则(Single responsibility principle)规定每个类都应该有
- POJO、VO和JavaBean区别和联系
fanmingxing
VOPOJOjavabean
POJO和JavaBean是我们常见的两个关键字,一般容易混淆,POJO全称是Plain Ordinary Java Object / Plain Old Java Object,中文可以翻译成:普通Java类,具有一部分getter/setter方法的那种类就可以称作POJO,但是JavaBean则比POJO复杂很多,JavaBean是一种组件技术,就好像你做了一个扳子,而这个扳子会在很多地方被
- SpringSecurity3.X--LDAP:AD配置
hanqunfeng
SpringSecurity
前面介绍过基于本地数据库验证的方式,参考http://hanqunfeng.iteye.com/blog/1155226,这里说一下如何修改为使用AD进行身份验证【只对用户名和密码进行验证,权限依旧存储在本地数据库中】。
将配置文件中的如下部分删除:
<!-- 认证管理器,使用自定义的UserDetailsService,并对密码采用md5加密-->
- mac mysql 修改密码
IXHONG
mysql
$ sudo /usr/local/mysql/bin/mysqld_safe –user=root & //启动MySQL(也可以通过偏好设置面板来启动)$ sudo /usr/local/mysql/bin/mysqladmin -uroot password yourpassword //设置MySQL密码(注意,这是第一次MySQL密码为空的时候的设置命令,如果是修改密码,还需在-
- 设计模式--抽象工厂模式
kerryg
设计模式
抽象工厂模式:
工厂模式有一个问题就是,类的创建依赖于工厂类,也就是说,如果想要拓展程序,必须对工厂类进行修改,这违背了闭包原则。我们采用抽象工厂模式,创建多个工厂类,这样一旦需要增加新的功能,直接增加新的工厂类就可以了,不需要修改之前的代码。
总结:这个模式的好处就是,如果想增加一个功能,就需要做一个实现类,
- 评"高中女生军训期跳楼”
nannan408
首先,先抛出我的观点,各位看官少点砖头。那就是,中国的差异化教育必须做起来。
孔圣人有云:有教无类。不同类型的人,都应该有对应的教育方法。目前中国的一体化教育,不知道已经扼杀了多少创造性人才。我们出不了爱迪生,出不了爱因斯坦,很大原因,是我们的培养思路错了,我们是第一要“顺从”。如果不顺从,我们的学校,就会用各种方法,罚站,罚写作业,各种罚。军
- scala如何读取和写入文件内容?
qindongliang1922
javajvmscala
直接看如下代码:
package file
import java.io.RandomAccessFile
import java.nio.charset.Charset
import scala.io.Source
import scala.reflect.io.{File, Path}
/**
* Created by qindongliang on 2015/
- C语言算法之百元买百鸡
qiufeihu
c算法
中国古代数学家张丘建在他的《算经》中提出了一个著名的“百钱买百鸡问题”,鸡翁一,值钱五,鸡母一,值钱三,鸡雏三,值钱一,百钱买百鸡,问翁,母,雏各几何?
代码如下:
#include <stdio.h>
int main()
{
int cock,hen,chick; /*定义变量为基本整型*/
for(coc
- Hadoop集群安全性:Hadoop中Namenode单点故障的解决方案及详细介绍AvatarNode
wyz2009107220
NameNode
正如大家所知,NameNode在Hadoop系统中存在单点故障问题,这个对于标榜高可用性的Hadoop来说一直是个软肋。本文讨论一下为了解决这个问题而存在的几个solution。
1. Secondary NameNode
原理:Secondary NN会定期的从NN中读取editlog,与自己存储的Image进行合并形成新的metadata image
优点:Hadoop较早的版本都自带,
 image
image
 image
image image
image image
image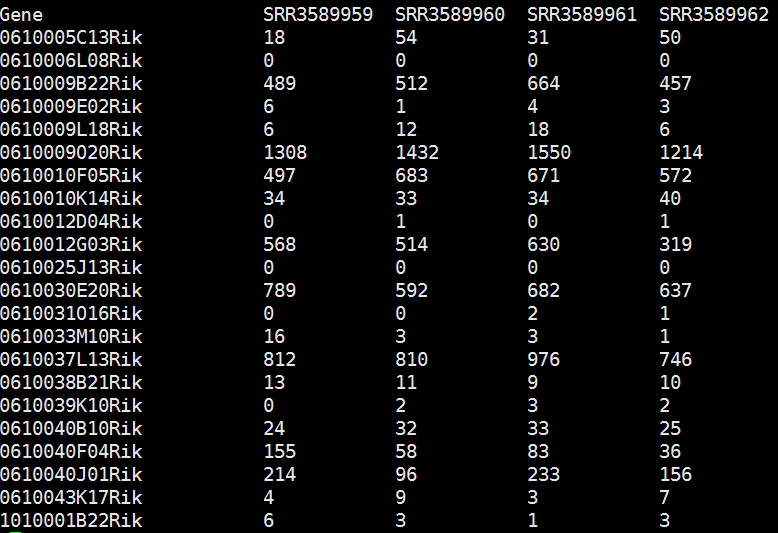 image
image