下游分析
下游分析可能是比较成熟化的流程分析,对服务器的要求比较高一些,下游分析则是对背景知识要求高一些,例如VCF的格式,遗传学的三大定律等。
让我们先安装并加载所需的R包
install.pacakges("devtools")
install.packages("vcfR")
devtools:install_github("xuzhougeng/binmapr")
library("vcfR")
library("binmapr")
然后我们需要利用vcfR读取VCF文件
vcf <- read.vcfR("04-variant-filter/snps.vcf")
接着从VCF对象中提取两个关键信息,AD(Allele Depth)和GT(Genotype)
gt <- extract.gt(vcf)
ad <- extract.gt(vcf, "AD")
gt是一个基因型矩阵,基于之的前过滤操作,所以这里只会有"0/0", "0/1","1/1"这三种情况,而ad则是等位基因的count数. 我们用head查看前10行来了解下情况,
> head(gt)
SRR6327815 SRR6327816 SRR6327817 SRR6327818
chr01_1151 "0/0" "1/1" "0/0" "0/1"
chr01_6918 "0/0" "1/1" "0/0" "0/1"
chr01_17263 "0/0" "1/1" "0/0" "0/1"
chr01_21546 "1/1" "1/1" "0/0" "0/1"
chr01_24732 "1/1" "0/0" "0/0" "0/0"
chr01_33667 "1/1" "1/1" "0/0" "0/1"
> head(ad)
SRR6327815 SRR6327816 SRR6327817 SRR6327818
chr01_1151 "14,0" "0,18" "10,0" "11,3"
chr01_6918 "31,0" "0,30" "32,0" "21,5"
chr01_17263 "46,0" "0,34" "20,0" "21,8"
chr01_21546 "0,25" "0,20" "17,0" "16,3"
chr01_24732 "0,29" "36,0" "21,0" "31,0"
chr01_33667 "0,26" "0,31" "28,0" "29,11"
其中SRR6327815, SRR6327816, SRR6327817, SRR6327818 分别对应着 KY131, DN422, T-pool 和 S-pool
仔细观察的话,你会发现一个chr01_21546是一个有趣的位置,因为双亲都是纯合情况下,SRR6327818居然是 "16,3", 基因型是"0/1", 这既有可能是亲本是杂合但没有测到,也有可能是后代测错了,一个简单粗暴的方法就是删掉它。此外我们还需要考虑是选择KY131是"1/1"且 "DN422" 是 "0/0"的位点。还是选择KY131是"0/0", 且 "DN422" 是 "1/1"位点。最好的方法就是两种都测试一下。
首先测试KY131是"1/1"且 "DN422" 是 "0/0"的位点
mask <- which(gt[,"SRR6327815"] == "1/1" & gt[,"SRR6327816"] == "0/0")
ad_flt <- ad[mask,c("SRR6327817", "SRR6327818")]
colnames(ad_flt) <- c("T_Pool", "S_Pool")
这一波过滤,我们从原来的80w位点中留下了20w个位点,因此测双亲很重要,能够极大的降低噪音。
然后,我们就可以根据AD计算SNP-index,即 ALT_COUNT / (REF_COUNT + ALT_COUNT), 在0-1之间。
freq <- calcFreqFromAd(ad_flt, min.depth = 10, max.depth = 100)
这里设了一个最大和最小深度用于计算频率,太大的深度可能是同源基因或者是重复序列,太低的深度在计算的时候不太准确。
freq2 <- freq[Matrix::rowSums(is.na(freq)) == 0, ]
接着我们就可以尝试去重现文章的结果了
par(mfrow = c(3,4))
for (i in paste0("chr", formatC(1:12, width = 2, flag=0)) ){
freq_flt <- freq2[grepl(i,row.names(freq2)), ]
pos <- as.numeric(substring(row.names(freq_flt), 7))
plot(pos, freq_flt[,2] - freq_flt[,1], ylim = c(-1,1),
pch = 20, cex = 0.2,
xlab = i,
ylab = expression(paste(Delta, " " ,"SNP index")))
}
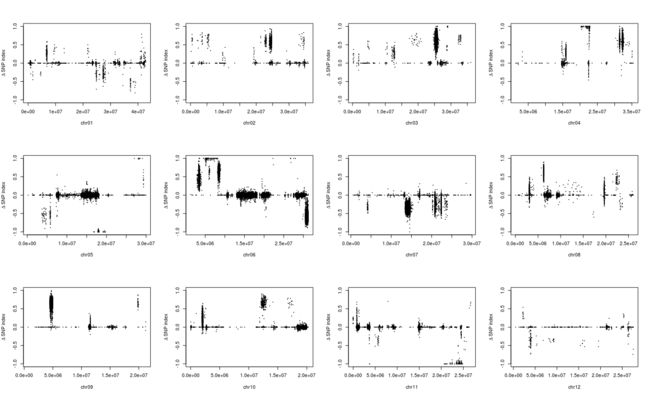
从图中,我惊讶的发现一些染色体上的部分区域居然都没有标记了,所以我们选择KY131是"0/0", 而 "DN422" 是 "1/1"的位点进行分析
mask <- which(gt[,"SRR6327815"] == "0/0" & gt[,"SRR6327816"] == "1/1")
ad_flt <- ad[mask,c("SRR6327817", "SRR6327818")]
colnames(ad_flt) <- c("T_Pool", "S_Pool")
freq <- calcFreqFromAd(ad_flt, min.depth = 10, max.depth = 100)
freq2 <- freq[Matrix::rowSums(is.na(freq)) == 0, ]
par(mfrow = c(3,4))
for (i in paste0("chr", formatC(1:12, width = 2, flag=0)) ){
freq_flt <- freq2[grepl(i,row.names(freq2)), ]
pos <- as.numeric(substring(row.names(freq_flt), 7))
plot(pos, freq_flt[,1] - freq_flt[,2], ylim = c(-1,1),
pch = 20, cex = 0.2,
xlab = i,
ylab = expression(paste(Delta, " " ,"SNP index")))
}
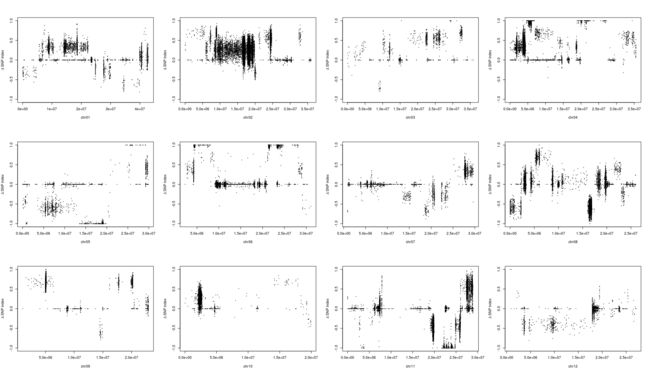
从结果中,我们能够发现一个有趣的结果,不同的筛选标记标准会导致标记在染色体上分布发生变化,这可能意味着在某种的筛选标准下,会使得和性状有关的位点明显的被过滤掉。
根据文章里的结果,候选基因落在6号染色体的20-25M中,也就是选择KY131是"0/0", 而 "DN422" 是 "1/1"的位点结果和原文比较类似。那么如果我们原本不知道这个结果应该怎么办?这其实也不是什么问题,像我这样把两幅图都做出来,然后和实验设计者交流下,也就差不多知道答案了。
除了SNP-index外,文章还有一个ED(Euclidean distance)方法用于定位,我根据文章的公式和自己的理解写了下代码
mask <- which(gt[,"SRR6327815"] == "0/0" & gt[,"SRR6327816"] == "1/1")
ad_flt <- ad[mask,c("SRR6327817", "SRR6327818")]
ED_list <- apply(ad_flt, 1, function(x){
count <- as.numeric(unlist(strsplit(x, ",",fixed = TRUE,useBytes = TRUE)))
depth1 <- count[1] + count[2]
depth2 <- count[3] + count[4]
ED <- sqrt((count[3] / depth2 - count[1] / depth1)^2 +
(count[4] / depth2- count[2] /depth1)^2)
return(ED^5)
})
par(mfrow = c(3,4))
for (i in paste0("chr", formatC(1:12, width = 2, flag=0)) ){
ED_flt <- ED_list[grepl(i,names(ED_list))]
pos <- as.numeric(substring(names(ED_flt), 7))
plot(pos, ED_flt,
pch = 20, cex = 0.2,
xlab = i,
ylab = "ED")
}

我复现的结果和文章的区别在于,少一个拟合线,也就是以1Mb为区间,每次滑动10Kb计算均值。这个部分稍微比较复杂,留到下一篇单独讲解。
版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。
