刘小泽写于18.12.31
再次知识迭代:打算以上中下三篇来认识一个新事物
上篇:主要了解VCF的背景知识;
一般我们会从WES的上游得到SNP、InDel等信息,这些重要的信息都保存在VCF中,那么怎么对这些变异进行提取、评估与解释呢?一起来学习一下
VCF(Variant Call Format)是什么?
之前也写过一篇相关的,这次想要更深层次去了解它https://www.jianshu.com/p/957efb50108f
我们知道,variant calling(找变异)的过程发生在alignment(比对)之后,那么肯定流程更加复杂,因此variant calling得到的结果也要更加精炼、内容更加丰富。于是VCF文件接手了这个棘手的工作。了解VCF,对于想要另辟蹊径发现新研究内容的人来说,真的是一块宝藏,就看你怎么挖掘了。
主要内容
得到一个VCF文件,首先看到的就是它的Header(表头),如下:(其实有非常非常多的头信息…这里只写几行)
##fileformat=VCFv4.1
##FILTER=
##FORMAT=
##FORMAT=
##GATKCommandLine.HaplotypeCaller=
##contig=
##reference=file:human_genome_b37.fasta
第一行是VCF的版本信息,看似没用,但实际上我们在分析其他数据时,并不能保证一直使用最新的VCF格式,因此检查下VCF版本确保后续提取正确
FILTER行是说过滤了什么内容;
FORMAT和INFO 相当于变异位点的注释信息;
CommandLine 是说使用的call variant工具信息;
contig和reference 是当重复别人数据时,恰好没告诉你数据来源,这时就可以参考这个
接下来才是重点:Records信息
包含至少8列tab分割的常规信息(用来描述变异位点),第9列及以后表示各个样本的变异信息(可以包括成百上千个样本)
前9列信息包括:
CHROM:变异发生的chromosome或者contig
POS:变异发生的基因组坐标(对缺失来讲,显示的是缺失开始的位置)
ID:一般是dbSNP的ID(可有可无)
-
REF:Forward strand(正链)上的参考等位基因
补充一下基础知识:
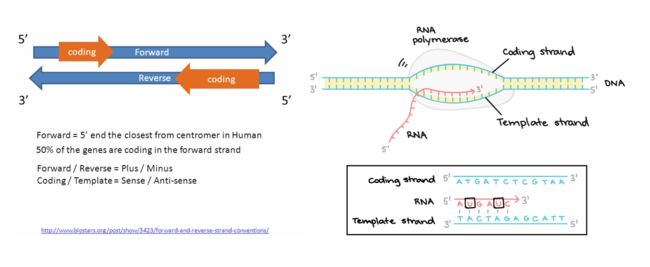 image.png
image.png等位基因(allele):一对同源染色体相同位置上控制某一性状的不同形态的基因,可以简单想成控制同一性状的不同基因
ALT:正链上对应的改变的等位基因(可以有多个)
QUAL:
REF/ALT变异位点存在的概率(和FASTQ的质量值、SAM的MAPQ一样,都是Phred值-10 * log(1-p))FILTER:数据一般都要经过适当的过滤后才能继续使用variant callset(变异位点数据集),关于是否完成过滤会给出三种说明:
一是给出没有通过过滤的变异位点;二是PASS表示全部通过了过滤;三是.表示这个位点没有任何过滤INFO:以
tag=value的形式给出位点注释信息;分号;分割FORMAT:针对样本的注释;冒号
:分割,并且对应后面各个sample中的信息
# 大体就是这样
#CHROM POS ID REF ALF QUAL FILTER INFO FORMAT align.bam
AF086833 60 . A T 54 . DP=43 GT:PL 0/1:51,0,48
上面是关于VCF的大体了解,下面具体看看重要的部分
具体--REF/ALT
REF表示在POS位置的参考等位基因;ALT表示在POS位置的所有变异
开始想象场景:我们用软件发现,在60bp处发现了一个变异,是A变成了T,那么应该这么表示:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT align.bam
AF086833 60 . A T 54 . DP=43 GT:PL 0/1:51,0,48
如果同一个位置检测到有两种可能发生的变异呢?
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT align.bam
AF086833 60 . A T,C 43.2 . DP=95 GT:PL 1/2:102,124,42,108,0,48
事情还没完,刚刚两个例子都只是一个变异位点,但实际上有可能多个位点发生缺失(而这才是VCF复杂的开始),例如:58、59、60碱基(GGA)发生了缺失
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT align.bam
AF086833 55 . TGAGGA TGA 182 . INDEL;IDV=42 GT:PL 0/1:215,0,255
AF086833 60 . A C,T 39.8 . DP=126 GT:PL 1/2:99,87,47,86,0,49
这里注意:虽然我们看到缺失是从58碱基开始发生的,但是记录的是从55碱基。你会不会好奇:为什么是55而不是准确的58?为什么要表示成TGAGGA->TGA,而不是GAGGA->GA或者GGA->空 呢?这个需要引入一个新词汇”variant normalizatoin“,也就是说,所有的结果展示都是有规定的。
variant normalization: 2015年Unified representation of genetic variants文章中就论述了这个一致性标记的问题,其中写道:
A genetic variant can be represented in the Variant Call Format (VCF) in multiple different ways. Inconsistent representation of variants between variant callers and analyses will magnify discrepancies between them and complicate variant filtering and duplicate removal.
于是制定了VCF的标准化,来方便交流,规定以下几点:
用尽可能少的字母来表示变异位点;
等位基因长度不为0;
变异向左”贪婪比对“ (也就是说:一直向左比对,直到不匹配为止,然后以最左边的碱基位置表示变异的起始位置)
https://academic.oup.com/bioinformatics/article/31/13/2202/196142
因此,这里写成TGAGGA->TGA就是表示缺失位点GGA向左最远可以匹配到第55号T碱基处
具体--FORMAT
假设得到的VCF中9-11列内容如下:
#FORMAT sample1 sample2
GT:PL 0/1:51,0,48 1/1:34,32,0
表示的意思就是:在样本1中发现的变异中含有GT=0/1和PL=51,0,48这样的信息,样本2中的变异含有GT=1/1和PL=34,32,0这样的信息
看到两个名词GT和PL,那么它们是什么意思呢?
我们可以回过头去Header部分看一看,将会找到如下内容:
##FORMAT=
##FORMAT=
还感觉看不太懂?不着急,往下接着学
具体--Genotypes
尽管我们可以根据REF和ALT知道了碱基发生了怎样的变化,但是我们想知道这个变化是只是发生在一个DNA拷贝中,还是两个拷贝都有?需要用一个指标来量化这种变异~基因型(Genotype)。GT就是用来表示样本中这个位点的基因型,其中0表示参考REF,1表示变异ALT的第一个entry,2表示ALT的第二个entry(以此类推)
对于二倍体生物,GT表示了一个样本中的等位基因:
-
0/0表示样本是纯合子,并且和参考的等位基因一样 -
0/1表示样本是杂合子,有一个参考的等位基因,一个变异的等位基因 -
1/2样本是杂合子,两个都是变异的等位基因 -
1/1样本是纯合子,且两个都是变异的第一个等位基因 -
2/2样本是纯合子,且两个都是变异的第二个等位基因(以此类推)
当然,如果不是二倍体,命名原理也是一样:单倍体(Haploid)只有一个GT值;多倍体有多个GT值
具体--Allele
A开头的大多和等位基因相关,如
- AC(Allele Count):该Allele数目
- AF(Allele Frequency):该Allele频率
- AN(Allel Number):Allele总数目
对于二倍体样本(Diploid sample),基因型为0/1表示杂合子:AC=1表示Allele数为1(即该位点只有1个等位基因发生变异);AF=0.5频率为0.5(该位点只有50%的等位基因发生变异);AN=2表示总Allele为2。
基因型为1/1表示纯合子,AC=2,AF=1,AN=2
正常人的二倍体基因组位点只有杂合和纯合两种情况,因此杂合AF一定是0.5,纯合AF一定是1,但实际的VCF数据是通过测序数据来的,随机性加上测序深度不够带来的系统误差,就使得结果变得不那么理想
具体--Genotype likelihoods
直白地说就是”基因型可能性“,就是用来衡量不同基因型可能发生的概率,这是利用p-value统计,因此0表示可能性最大,例如:
GT:PL 0/1:51,0,48
其中PL这一项有三个数值,分别对应三种可能的基因型(0/0,0/1,1/1)发生的概率:第一个数值51表示基因型为0/0的概率是Phred值51 ,也就是1x10^6 ;第二个数值0表示基因型为0/1的概率是0(和GT判断的一致);第三个数值48表示基因型为1/1的概率是1x10^5
具体--Allele depth and depth of coverage
软件判断是那种基因型,到底是不是发生了变异,是需要一定的统计方法的,主体就是之前比对的结果BAM文件,其中包含了reads的比对信息,这里就是根据reads比对的数量进行判断
- AD(DepthPerAlleleBySample): unfiltered allele depth 就是有多少reads出现了某个等位基因(其中也包含了没有经过variant caller过滤的reads),但是不包括没意义的reads(就是那些统计结果不过关的,没法说服软件相信这个等位基因)。在二倍体样本中表示为:逗号分隔的两个值,前者为对应的ref基因型,后者为对应alt的基因型
- DP(Coverage,即reads覆盖度,指一些reads被过滤后的覆盖度): filtered depth, at the sample level 只有通过variant caller软件过滤后的reads才能计算入内,但是DP也纳入了那些经过过滤但没有意义的reads(uninformative reads),这一点又和AD不同
具体--Genotype Quality
GQ就是用Phred值来表示GT判断的准确性,它和PL相似,但是取值不同。PL最小值0表示最准确,GQ一般取PL的第二个小的值(除非第二小的PL大于99)。在GATK分析中,GQ最大就限制在99,因为超过99其实是没有什么意义的,并且还多占字符。因此,如果GATK中发现PL值中第二个小的值比99还要大,软件就将GQ标为99。用GQ值就可以得到,第一位和第二位之间到底差了多少,因此可以快速判断分析的准不准,选择第一个靠不靠谱
Basically the GQ gives you the difference between the likelihoods of the two most likely genotypes. If it is low, there is not much confidence in the genotype.
做一个小总结--VCF帮助判断基因型
现在来看看NA12878基因(1: 899282)的统计情况
1 899282 rs28548431 C T [CLIPPED] GT:AD:DP:GQ:PL 0/1:1,3:4:26:103,0,26
这个位点的GT=0/1,可以判断基因型是C/T;GQ=26表示排名第二基因型的可能性是0.0025,结果不是很好,因为虽然判断的第一位基因型的PL 为0很可靠,但是毕竟相差不多,很难推翻第二位(如果GQ值再大一些,我们就更有信心说明判断的C/T基因型是正确的)。当然,这里GQ的原因很有可能是取样太少,只有4条reads在这个位点作为参考(DP=4),这四条中有1条带参考的碱基信息,另外3条与参考不一致,存在变异(AD=1,3)
因此,重点的结论来啦!尽管我们相信,这个位点确实存在变异,但是假阳性依然存在,也就意味着基因型判断结果不一定是杂合子C/T,有一定的可能是变异纯合T/T(PL(1/1)=26),但一定不可能是参考纯合C/C (PL(0/0)=103)
参考:
VCF short summary:http://www.htslib.org/doc/vcf.html
VCF Poster:http://vcftools.sourceforge.net/VCF-poster.pdf
VCF 说明书: http://samtools.github.io/hts-specs/
GATK解释VCF:https://gatkforums.broadinstitute.org/gatk/discussion/1268/what-is-a-vcf-and-how-should-i-interpret-it
Difference between QUAL and GQ annotations https://software.broadinstitute.org/gatk/documentation/article.php?id=4860
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
