生信星球狗粮特供:
我们的公众号生信星球的开始,就是我正式开始学生信的时候,零星时间看过的东西都会忘,只有找个办法push自己,把学到的东西系统性输出,才能短期得到提高,公众号就是一个非常好的选择。每天都发, 一天一篇,我们两个人所以两天一篇,这个节奏就非常好,在别人眼里可能这是一个坚持的理由,但在我们眼里,那不叫坚持,这是两个人的奋斗记录,是最好的回忆啊,当然是动力十足的。所以谁也不肯拖后腿,一起进步,我们还从一开始就记下了天数,这是一个多好的爱情故事哈哈哈哈。
9个月前开始学生信,我还处于看不懂大神发过来的任务,一脸懵逼等着豆豆拯救的状态,但是那时候就敢写了,为了写很短的一篇,要花好长时间理解,反复测试写的对不对。真正的大神都是实战中练出来的,可我一个路边捡来的做蛋白的小玩家,生信是什么完全没听过,哪来的实战。。。
那时候就见过了这20题,蒙蒙的不会做,做了5道题就败下阵来。后来接触到了R语言,感觉比linux简单多了,半年业余时间全都贡献给R语言了,甚至学了两个月的时候和豆豆一起开了学习小组,开始写非常详细的教程给别人讲,大概是因为与生俱来的感觉,我就喜欢讲课,上大学时想报师范的,因缘际会去学了农学,绕了一圈还是当老师。因为长时间用不到,所以再也不记得linux了。
这几天来到珠海参加生信技能树的线下培训,第一天当老师讲R语言,第二天当学生学linux。听完老师现场授课,有一种奇经八脉被打通的感觉,linux和R语言根本就是互通的呀。晚上作业是做20道题。当我只觉得眼熟,直到我搜答案的时候搜到了我自己写的哈哈哈哈哈哈哈哈哈这事够我笑一年。
我学到现在略有小小小小小成,是非常幸运的,毕竟我有一个大神男票,还有一个大神老板啊,我膨胀。虽然现在还是只会R语言的状态,但我马上毕业了,有这么好的资源一定进步很快,2019我要加油。
言归正传,做题看答案咯!如果有误请指出呀。
题目链接:http://www.bio-info-trainee.com/2900.html
一、在任意文件夹下面创建形如 1/2/3/4/5/6/7/8/9 格式的文件夹系列。

考察新建层级目录。
mkdir -p
二、在创建好的文件夹下面,比如我的是 /Users/jimmy/tmp/1/2/3/4/5/6/7/8/9 ,里面创建文本文件 me.txt

考察新建文本文档。
touch me.txt
三、在文本文件 me.txt 里面输入内容:
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
老师推荐使用cat > 来重定向。
前三题效果如下:

删除上面创建的文件夹 1/2/3/4/5/6/7/8/9 及文本文件 me.txt

五、在任意文件夹下面创建 folder1~5这5个文件夹,然后每个文件夹下面继续创建 folder1~5这5个文件夹,效果如下:
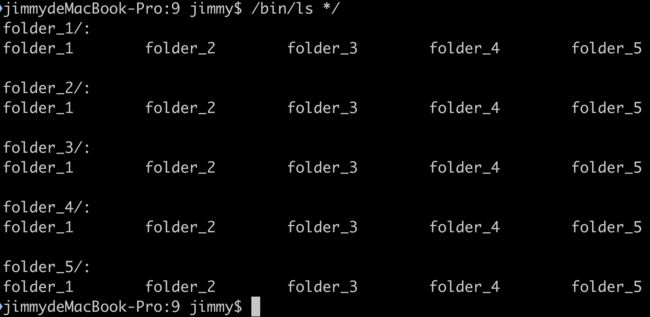
这个没思路,选择了搜索

看了第一篇 在它的命令上面修改:
mkdir -p /tmp/test/{1..9}/src
改成了
mkdir -p folder{1..5}/folder{1..5},一次成功的我膨胀起来。

六、在第五题创建的每一个文件夹下面都 创建第二题文本文件 me.txt ,内容也要一样。
还是不会,去搜索,结果发生了一件非常尴尬的事情,我搜到了半年前自己写的答案,还是盗版的哈哈哈哈哈哈哈哈哈哈哈哈哈哈哈哈哈哈哈
https://cloud.tencent.com/developer/news/231651
所以答案是
方法一
for dirs in folder{1..5}/folder{1..5}; do cp me.txt $dirs; done

方法二
用echo和xargs 天呐,我忘了这两个方法哪个是我写的了,震惊。这么难,半年前的我是怎么搞明白的?那时候应该想不到再次见到这个题的时候我已经成了R语言讲师吧。
echo folder{1..5}/folder{1..5} | xargs -n 1 cp -v me.txt

七,再次删除掉前面几个步骤建立的文件夹及文件
考察批量删除嘛,老师说删除之前ls先看一下
ls folder*
rm -rf folder*
ls
八、下载 http://www.biotrainee.com/jmzeng/igv/test.bed 文件,后在里面选择含有 H3K4me3 的那一行是第几行,该文件总共有几行。
下载是wget -c http://www.biotrainee.com/jmzeng/igv/test.bed
-c代表的是断点续传。
按行查找所以应该是用grep,找了一下白天老师讲的。https://www.jianshu.com/p/22a4324ddfdf
有感觉了,搞起来!
nl test.bed |grep H3K4me3
nl test.bed |wc -l

九、下载 http://www.biotrainee.com/jmzeng/rmDuplicate.zip 文件,并且解压,查看里面的文件夹结构
wget -c http://www.biotrainee.com/jmzeng/rmDuplicate.zip
unzip rmDuplicate.zip
tree rmDuplicate
十、打开第九题解压的文件,进入 rmDuplicate/samtools/single 文件夹里面,查看后缀为 .sam 的文件,搞清楚 生物信息学里面的SAM/BAM 定义是什么。
cd rmDuplicate/samtools/single
ls
#tmp.sam
尝试了cat less等等发现好乱,就去找怎么样好好显示,最后找到了less -S这个命令!显示效果好多了。
这个定义,谷歌就好了
找到的链接就看:https://www.jianshu.com/p/9c99e09630da
十一、安装 samtools 软件
今天还没有讲conda安装,不过有点印象,又去搜教程,,额又发现我们写过。https://mp.weixin.qq.com/s/FBsY8hRjTS6ih2RvY47I6Q
先安装conda,再用conda安samtools
wget -c https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
#全部yes,enter
source ~/.bashrc
conda install samtools
十二、打开 后缀为BAM 的文件,找到产生该文件的命令。 提示一下命令是:
/home/jianmingzeng/biosoft/bowtie/bowtie2-2.2.9/bowtie2-align-s --wrapper basic-0 -p 20 -x /home/jianmingzeng/reference/index/bowtie/hg38 -S /home/jianmingzeng/data/public/allMouse/alignment/WT_rep2_Input.sam -U /tmp/41440.unp
先要找bam后缀的文件,只能记得找文件命令是find,但是参数不记得。所以搜索

所以看参数,想起来了,按照name查找:
find rmDuplicate/ -name *.bam
找到产生它的命令,记得n久前老板说过是在bam的头文件中有,但下载的bam文件好奇怪看不懂

这个跳过。
十三题、根据上面的命令,找到我使用的参考基因组 /home/jianmingzeng/reference/index/bowtie/hg38 具体有多少条染色体。
我非常机智的在教学服务器的teach文件夹下找到了hg38.fa!
/teach/database/genome
可以看到这个是fa格式,所以要找找有多少>开头的行,然后去重复。
less -S hg38.fa |grep '^>' |sort |uniq -c
less -S hg38.fa |grep '^>' |sort |uniq -c |wc -l
#返回结果是400多行

虽然不太对劲。。。除了染色体还有些别的名称返回,但方法搞会了。
十四题、上面的后缀为BAM 的文件的第二列,只有 0 和 16 两个数字,用 cut/sort/uniq等命令统计它们的个数。
我的bam文件不对劲aaa。
切割第二列然后去重统计呗?我会。
cut -f 2 x.bam |sort |uniq -c
应该对吧。
十五题、重新打开 rmDuplicate/samtools/paired 文件夹下面的后缀为BAM 的文件,再次查看第二列,并且统计
怕了怕了,打不开都是乱码。
十六题、下载 http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip 文件,并且解压,查看里面的文件夹结构, 这个文件有2.3M,注意留心下载时间及下载速度。
下载速度100k吧还可以。
wget -c http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip
unzip sickle-results.zip
tree sickle-results

十七题、解压 sickle-results/single_tmp_fastqc.zip 文件,并且进入解压后的文件夹,找到 fastqc_data.txt 文件,并且搜索该文本文件以 >>开头的有多少行?
unzip sickle-results/single_tmp_fastqc.zip
cd single_tmp_fastqc/
less -S fastqc_data.txt |grep '^>>'|wc -l
十八题、下载 http://www.biotrainee.com/jmzeng/tmp/hg38.tss 文件,去NCBI找到TP53/BRCA1等自己感兴趣的基因对应的 refseq数据库 ID,然后找到它们的hg38.tss 文件的哪一行。
https://www.ncbi.nlm.nih.gov/gene/7157
wget -c http://www.biotrainee.com/jmzeng/tmp/hg38.tss
#写了一个这个命令,觉得没问题可是没返回值
less -S hg38.tss |grep -n 'TP53'
head hg38.tss #一看 这里面不是基因名而是某个基因id。
#所以需要一个id转换,算了麻烦,随便找一个嘻嘻
nl hg38.tss |grep -n 'NR_028459'
十九题、解析hg38.tss 文件,统计每条染色体的基因个数。
文件比较简单,也就是统计第二列的重复值情况就好。切片排序去重一步到位!
cut -f 2 hg38.tss |sort|uniq -c
二十题、解析hg38.tss 文件,统计NM和NR开头的熟练,了解NM和NR开头的含义。
easy!hhh
less -S hg38.tss |grep '^NM'|wc -l
less -S hg38.tss |grep '^NR'|wc -l
NM_:mRNA mixed,转录组产物序列;成熟mRNA转录本序列;
NR_:RNA mixed,非编码的转录子序列,包括结构RNAs,假基因转子等;
搜索过程

找到的链接:http://blog.sina.com.cn/s/blog_72512a1d0102xp6f.html