一、测序数据的格式转换
sra文件下载好后,使用fastq-dump转换数据格式:
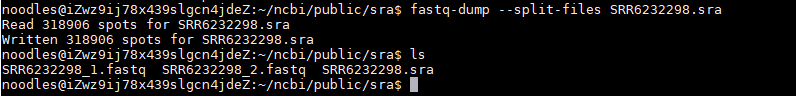
fastq-dump --split-files SRR6232298.sra
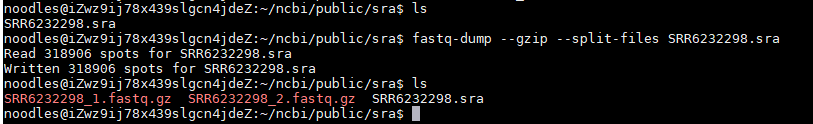
fastq-dump --gzip --split-files SRR6232298.sra
#转换格式的同时解压为gz文件,节省空间
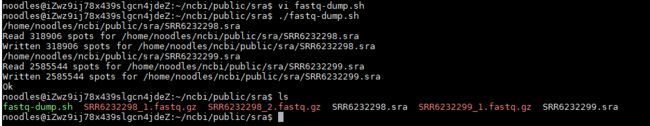
当有多个sra文件时,可通过脚本进行批量解压:
vi fastq-dump.sh #创建一个脚本文件,内容如下:
#!/bin/bash
for i in ~ncbi/public/sra/sra*
do
echo $i
fastq-dump --gzip --split-files $i
done
echo OK
二、测序数据的质控
FastQC---测序数据质控的软件
是一个java软件,下载后可以直接使用(免安装编译),但是需要自行配置好java环境
首先我们配置java环境(已下好java文件,为下述的jdk-8u172-linux-x64.tar.gz):
sudo mkdir /usr/java
sudo tar -zvxf /home/noodles/Biosofts/jdk-8u172-linux-x64.tar.gz -C /usr/java/
sudo cd /usr/java
sudo ln -s jdk1.8.0_172 latest
sudo ln -s /usr/java/latest default
sudo vi /etc/profile
末尾加上如下三行:
export JAVA_HOME=/usr/java/latest
export PATH=$JAVA_HOME/bin:$JAVA_HOME/jre/bin:$PATH
export CLASSPATH=.:$JAVA_HOME/lib/dt.jar:$JAVA_HOME/lib/tools.jar
source /etc/profile
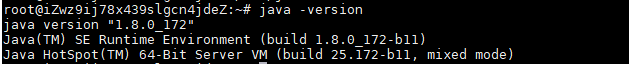
java -version
配置好java环境后,接着可以开始下载安装FastQC软件了
1. wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
2. unzip /home/noodles/Biosofts/fastQC/fastqc_v0.11.7.zip -d ~/Biosofts/fastQC
3. ~/Biosofts/fastQC/FastQC/fastqc -h
当我运行这步后,系统提示fastqc还未安装,通过apt install fastqc来安装,当进行安装后报错:

呈上:
4. 将fastqc加入环境变量:
echo 'export PATH=~/Biosofts/fastQC/FastQC:$PATH' >>~/.bashrc
source ~/.bashrc
fastqc -h
至此,已装好了fastQC软件,fastQC的使用方法如下:
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
各参数的含义:
-o --outdir 生成的报告文件的储存路径,生成的报告的文件 名是根据输入来定的
--extract 生成的报告默认会打包成1个压缩文件,使用这个 参数是让程序不打包
-t --threads 选择程序运行的线程数
-c --contaminants 污染序列选项,输入的是一个文件,格式 是Name [Tab] Sequence,里面是可能的污染序列
-a --adapters 也是输入一个文件,文件的格式Name [Tab] Sequence,储存的是测序的adpater序列信息;默认已有一些
-q --quiet 安静运行模式,一般不选这个选项的时候,程序 会实时报告运行的状况。
接下来以用fastqc处理之前下载的seq为例,当我处理SRR6232298_1.fastq.gz时报错:
经过检查后原来是fastqc的权限问题,增加权限即可:
三、测序数据过滤
数据过滤软件用来切除接头序列和低质量碱基,目前也已有很多工具:Trimmomatic、seqtk、 cutadapt、 bbduk(BBmap). 下面以Trimmomatic为例介绍:
Trimmomatic 是一个广受欢迎的 Illumina 平台数据过滤工具。 Trimmomatic 支持多线程,处理数据速度快,主要用来去除 Illumina 平台的 Fastq 序列中的接头,并根据碱基质量值对 Fastq 进行修剪。软件有两种过滤模式,分别对应 SE 和 PE 测序数据,同时支持 gzip 和 bzip2 压缩文件。另外也支持 phred-33 和 phred-64 格式互相转化,不过现在 绝大部分 Illumina 平台的产出数据也都转为使用 phred-33 格式了 。
下载:
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.38.zip
安装:
unzip Trimmomatic-0.38.zip -d ~/Biosofts/trimmomatic/Trimmomatic038/
运行:
java -jar ~/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/trimmomatic-0.38.jar
添加环境变量:
echo 'export PATH=~/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/trimmomatic-0.38.jar:$PATH' >>~/.bashrc
source ~/.bashrc
然后以之前下载的SRR6232298_1.fastq.gz数据为例进行操作:
java -jar ~/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/trimmomatic-0.38.jar PE -phred33 SRR6232298_1.fastq.gz ./trim_out/output_forward_paired.fq.gz ./trim_out/output_forward_unpaired.fq.gz ./trim_out/output_reverse_paired.fq.gz ./trim_out/output_reverse_unpaired.fq.gz ILLUMINACLIP:/home/noodles/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/adapters/TruSeq2-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
(上述命令中,output_forward_paired.fq.gz、output_forward_unpaired.fq.gz、output_reverse_paired.fq.gz、output_reverse_unpaired.fq.gz四个文件由自己命名,软件自行生成。)结果报错:
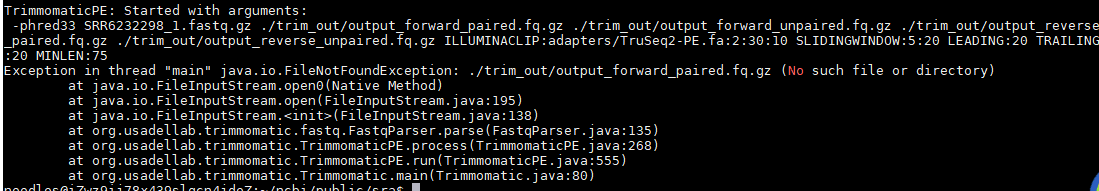
经仔细检查后,发现问题出在下面的点:
trimmomatic有两种模式即SE模式和PE模式,分别对应单末端测序模式和双末端测序模式,在 SE 模式下,只有一个输入文件和一个过滤之后的输出文件;在 PE 模式下,有两个输入文件,正向测序序列和反向测序序列,但是过滤之后输出文件有四个,过滤之后双端序列都保留的就是 paired,反之如果其中一端序列过滤之后被丢弃了另一端序列保留下来了就是 unpaired 。上述的问题就在于选用的是PE模式,但只输入了一个文件,所以报错。(SRR6232298.sra为双末端测序的数据,SRR6232298_1.fastq.gz和SRR6232298_2.fastq.gz为使用fastq-dump对SRR6232298.sra处理后所得,分别为正向测序序列和反向测序序列)因此当我将SRR6232298_1.fastq.gz和SRR6232298_2.fastq.gz都输入时,软件可正常运行:
java -jar ~/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/trimmomatic-0.38.jar PE -phred33 SRR6232298_1.fastq.gz SRR6232298_2.fastq.gz ./trim_out/output_forward_paired.fq.gz ./trim_out/output_forward_unpaired.fq.gz ./trim_out/output_reverse_paired.fq.gz ./trim_out/output_reverse_unpaired.fq.gz ILLUMINACLIP:/home/noodles/Biosofts/trimmomatic/Trimmomatic038/Trimmomatic-0.38/adapters/TruSeq2-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
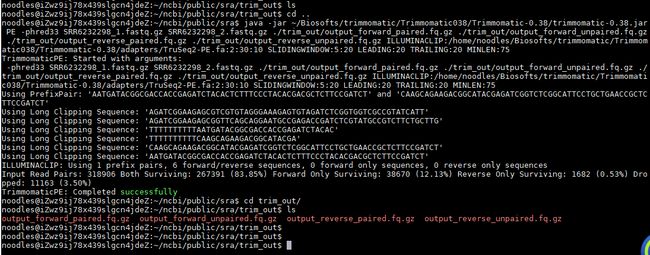
Trimmomatic 过滤数据的步骤与命令行中过滤参数的顺序有关,通常的过滤步骤如下:
1. ILLUMINACLIP: 过滤 reads 中的 Illumina 测序接头和引物序列,并决定是否去除反向互补的 R1/R2 中的 R2。
2. SLIDINGWINDOW: 从 reads 的 5' 端开始,进行滑窗质量过滤,切掉碱基质量平均值低于阈值的滑窗。
3. MAXINFO: 一个自动调整的过滤选项,在保证 reads 长度的情况下尽量降低测序错误率,最大化 reads 的使用价值。
4. LEADING: 从 reads 的开头切除质量值低于阈值的碱基。
5. TRAILING: 从 reads 的末尾开始切除质量值低于阈值的碱基。
6. CROP: 从 reads 的末尾切掉部分碱基使得 reads 达到指定长度。
7. HEADCROP: 从 reads 的开头切掉指定数量的碱基。
8. MINLEN: 如果经过剪切后 reads 的长度低于阈值则丢弃这条 reads。
9. AVGQUAL: 如果 reads 的平均碱基质量值低于阈值则丢弃这条 reads。
10. TOPHRED33: 将 reads 的碱基质量值体系转为 phred-33。
11. TOPHRED64: 将 reads 的碱基质量值体系转为 phred-64。
参考:NGS 数据过滤之 Trimmomatic 详细说明
下面两张图分别为未经Trimmomatic过滤和经过Trimmomatic过滤的SRR6232298_1.fastq.gz,经过fastQC质控后的结果:

