- 线程池创建及参数设置
运筹帷幄小红花
java开发语言后端
一、创建线程池以及线程池的各种参数分析://不指定最大线程数,默认是Int的最大值ExecutorServiceexecutorService=Executors.newCachedThreadPool();executorService.submit(()->{System.out.println("新建线程池对象,执行第一个默认线程");});executorService.shutdown(
- 【Bluedroid】HFP连接流程源码分析(一)
byte轻骑兵
解读AndroidjavaC++Android
Bluedroid蓝牙HFP(HFP,Hands-FreeProfile)连接流程涵盖多个环节,从前期准备到连接建立、状态管理以及维护与断开,各环节紧密相扣,确保蓝牙免提连接稳定可靠。一、概述1.1.连接前准备用户操作:用户需在Android设备上开启蓝牙功能。同时,目标蓝牙设备(如车载蓝牙)要进入配对模式,Android设备通过搜索发现目标设备并完成配对,此过程可能需用户输入PIN码或确认配对请
- unity游戏引擎架构设计分析
你一身傲骨怎能输
游戏引擎游戏引擎unity
Unity游戏引擎的架构设计是一个高度复杂且模块化的系统,它允许开发者创建跨多个平台的游戏和应用程序。以下是对Unity游戏引擎架构设计的分析:1.总体架构Unity引擎的总体架构可以分为几个主要层次:核心层(CoreLayer):这是引擎的基础,包含基本的数据类型、内存管理、线程和同步机制等。平台抽象层(PlatformAbstractionLayer):这一层负责处理不同平台的差异,确保游戏可
- 3D UNet和Swin-UNETR
学無芷境
计算机视觉
3DUNet和Swin-UNETR都是用于医学图像分析的深度学习网络,它们对三维(3D)数据进行特征提取和分割。3DUNet3DUNet是UNet架构的一个变体,专门设计用于处理三维医学图像数据。UNet最初是为二维(2D)图像分割任务设计的,具有典型的编码器-解码器结构。3DUNet扩展了这种架构,以便更好地处理具有深度信息的体积数据,如CT或MRI扫描。主要特点:编码器:逐渐下采样图像,提取并
- 推荐3D UNet实现:深度学习3D体素数据语义分割的利器!
滑辰煦Marc
推荐3DUNet实现:深度学习3D体素数据语义分割的利器!去发现同类优质开源项目:https://gitcode.com/在这个快速发展的深度学习时代,3DUNet已经成为3D图像处理领域中不可或缺的工具,尤其在医疗影像分析和3D物体识别等任务上展现出强大的潜力。这个开源项目为我们提供了一个高效、灵活的3DUNet实现,支持Tensorflow、PyTorch和Chainer三种主流深度学习框架。
- 2025 年最好的谷歌地图数据采集软件推荐
后端
2024年,谷歌地图抓取已成为企业和研究人员的一大变革。凭借不断更新的餐厅、酒店、药店等数据库,谷歌地图提供了大量信息。通过使用正确的谷歌地图抓取工具,您可以解锁有价值的见解,以进行潜在客户生成、市场研究和竞争分析。最好的谷歌地图抓取工具使您能够自动收集数据,节省时间和资源,同时确保准确性。无论您是想分析竞争对手还是扩大客户群,这些工具都能为您提供成功所需的优势。关键要点谷歌地图抓取工具可以访问有
- 如何理解 Tailwind CSS 的“功能类(Utility-first)”思想?
TailwindCSS是一个基于“功能类(Utility-first)”思想的CSS框架。它与传统的CSS框架(如Bootstrap或Foundation)有很大的不同,不是通过预定义的组件来构建UI,而是通过一系列低级的、原子化的类来构建定制化的设计。这种方法使得开发者可以更加灵活、高效地定制网页布局和样式。在本文中,我们将深入探讨TailwindCSS的核心思想——功能类,并分析它如何帮助开发
- 目前主流游戏引擎的分析报告
游戏开发88
cocoscreatorunity游戏引擎
前言游戏引擎之争就像编程语言之争一样,在游戏开发圈永远是一个火爆的话题,目前市面上主流的一些游戏引擎,我们来给他们做一些比较,了解他们的历史,特点,为了严谨,备注一下写这个文章的时间编写时间是2021年4月20日。目前国内主流在用的游戏引擎有,Unity,Cocos,Laya,UE4,白鹭,接下来我们一起来分析这些引擎的特点。1:国民3D引擎UnityUnity,使用C#或Lua语言开发。国民3D
- 百万架构师第二十二课:源码分析:Spring 源码分析:Spring经典面试答疑|JavaGuide
后端
Spring面试解答上半节:面试中需要注意的细节动脑子,面试是一种交流面试的时候,要用心去感受当时面试场景了解自己,自己的长处、自己的短处(巧妙地扬长避短)了解1.公司的业务场景2.你是去面试什么岗位的?Java高级工程师实际工作经验是1年(如实填写)1、请描述SpringIOC的工作原理答:定位加载注册BeanFactoryBeanDefintion...1-3年1+ApplicationCon
- MATLAB语言的计算机基础
疯狂小小小码农
包罗万象golang开发语言后端
MATLAB语言的计算机基础引言在当今信息技术飞速发展的时代,编程能力已成为当代人士必备的一项基本技能。MATLAB(矩阵实验室)作为一种高级编程语言和环境,广泛应用于数据分析、算法开发、模型创建、数字图像处理和计算机视觉等多个领域。MATLAB以其强大的矩阵运算和可视化能力,成为了科研人员和工程师的重要工具,尤其在数学、物理、工程等学科中,它的应用不可或缺。本文将从MATLAB的基本概念、环境搭
- Python 爬虫实战案例 - 获取社交平台事件热度并进行影响分析
西攻城狮北
Python实用案例python爬虫事件热度影响分析
目录一、引言二、数据爬取三、数据分析四、可视化展示五、总结一、引言在当今信息爆炸的时代,社交平台成为了各类事件发酵和传播的重要场所。了解社交平台上事件的热度以及其潜在影响,对于舆情监测、市场营销、社会趋势分析等领域具有重要意义。本文将通过一个实际案例,展示如何使用Python爬虫技术获取社交平台上特定事件的相关数据,并对其热度和影响进行深入分析。在本篇博客中,我们将学习如何使用Python编写一个
- IPD新产品立项管理的典型问题分析(下)
汉捷咨询
ipd
其次是新产品立项缺乏规划,前瞻性不足。凡事预则立,不预则废,新产品的立项和开发同样需要预先规划,即产品规划。在这里提及的产品规划,一般指的是企业未来3~5年的产品规划,有企业把年度产品开发计划称为产品规划,这其实是不准确的。纵观国内大部分企业,真正把产品规划做起来的企业占比非常低,从数据来看低于10%。企业拿年度产品开发计划作为立项计划,会因为时间的关系,给立项的时间明显不足。“立项时间只给一个月
- 集团公司L1-L5级流程框架方法论:(1)L1级流程:为业务价值链,是业务流程的主干; (2)L2级流程:为运作模式层面的业务子流程,因场景不同而差异化; (3)L3级流程:为实现运营模式所需的业
公众号:优享智库
数字化转型数据治理主数据数据仓库大数据
集团公司L1-L5级流程框架方法论集团公司L1-L5级流程框架方法论L1级流程:业务价值链定义与作用构成要素与其他流程关系案例分析L2级流程:运作模式层面业务子流程场景差异化原因子流程分类与特点跨场景协同策略案例分析L3级流程:业务能力与业务活动业务能力定义及要求业务活动类型与目的与IT系统关系澄清案例分析L4级流程:业务与IT系统交互过程/工作流交互过程描述方法工作流设计原则常见问题与解决方案案
- 企业架构业务流程设计五步法
公众号:优享智库
数字化转型数据治理主数据数据仓库架构
企业架构业务流程设计五步法企业架构与业务流程概述企业架构定义及重要性业务流程概念及作用企业架构与业务流程关系第一步:明确战略目标与需求确定企业战略目标分析业务需求及痛点制定项目目标与计划第二步:梳理现有业务流程调研现有业务流程情况识别关键业务环节及问题评估现有流程效率与效果第三步:设计优化方案与实施路径制定优化策略及原则设计新业务流程框架图规划实施步骤与时间表第四步:落地实施与持续改进搭建支持新流
- 给Wordpress添加评分功能到评论表单
鱼仰泳
WordPress开发手记WordPressPHPcss前端网站开发
今天要给你的Wordpress添加评分功能到评论表单吗?评分功能效果图什么类型的网站需要评分?资源站教程站其他,我也没想到。。。但我这个网站,因为是电影类的网站,好像还是有点需要的,所以,我就给它加上。修改后台代码(functions.php)添加评分代码首先,你需要将下面代码复制到functions.php中://添加打分脚本到评论表单//codebyyangjiyongVX:uu0216fun
- 代码结构与模块化设计:Python 项目架构与高效开发技巧
全栈探索者chen
pythonpython架构开发语言模块化性能优化程序人生案例分析
代码结构与模块化设计:Python项目架构与高效开发技巧目录为什么模块化设计是高效开发的基础Python项目的理想目录结构模块与包:概念与使用详解模块化设计的核心原则常见设计模式与模块化案例分析:从零搭建模块化Python项目高级技巧:动态模块加载与插件化设计模块化开发中的常见问题与解决方案总结与实践建议1.为什么模块化设计是高效开发的基础模块化设计是一种将复杂的软件系统分解为多个小模块的开发方式
- 大数据毕业设计—基于python+Django自然灾害频发地区情况数据分析系统
qq_1406299528
python计算机毕业设计python大数据课程设计
一、项目技术开发语言:Pythonpython框架:Django软件版本:python3.7/python3.8数据库:mysql5.7或更高版本数据库工具:Navicat11开发软件:PyCharm/vscode前端框架:vue.js二、项目内容和项目介绍 1.项目内容 1.开发语言:该系统采用Python作为开发语言,Python具有优雅的语法和动态类型,以及解释型语言的本质,使其成为许多
- EOF分析在Python中的利器:eofs库使用指南
潘妙霞
EOF分析在Python中的利器:eofs库使用指南项目地址:https://gitcode.com/gh_mirrors/eo/eofs项目介绍eofs是一个专为Python环境设计的开源包,用于执行经验正交分解(EmpiricalOrthogonalFunction,EOF)分析。该库遵循GNUGPLv3许可协议,旨在简化Python中进行EOF分析的流程。它特别适合处理大型时空数据集,通过高
- 利用Python爬虫获取阿里巴巴商品详情:代码示例与实践指南
小爬虫程序猿
APIpython爬虫开发语言
在电商数据分析和市场研究中,获取商品详情是至关重要的一步。虽然阿里巴巴开放平台提供了官方API来获取商品信息,但在某些情况下,使用爬虫技术来抓取数据也是一种有效的手段。本文将介绍如何利用Python爬虫获取阿里巴巴商品详情,并提供详细的代码示例。一、准备工作(一)环境搭建确保你的Python环境已经安装了以下必要的库:requests:用于发送HTTP请求。BeautifulSoup:用于解析HT
- 阿里巴巴商品搜索结果页结构复杂吗?
小爬虫程序猿
Javapython爬虫信息可视化
阿里巴巴商品搜索结果页的结构相对复杂,但通过合理的方法和工具,可以有效地解析和提取所需信息。以下是对阿里巴巴商品搜索结果页结构的分析:一、返回值的主要组成部分(一)商品总数(totalItems)表示搜索到的商品总数,这一数值直接反映了搜索结果的规模。电商平台可以根据商品总数判断是否需要进行分页处理,以优化用户体验。(二)商品列表(items)包含与搜索关键字匹配的商品列表,每个商品都是一个对象,
- Rabbitmq源码分析,重复消费问题的redis或数据库代码实现
xweiran
rabbitmq分布式java架构jvm数据结构后端
目录底层源码解析自定义唯一id算法MessageProperties类的相关实现自定义消息ID生成器配置和使用Rabbitmq是怎么判断是不是重复消息的呢?通过Redis的幂等性处理消息消费者实现分布式锁实现的重复检测完整的消息处理流程基于数据库实现Mapper接口消息处理服务RabbitMQ消息消费者底层源码解析RabbitMQ判断重复消息主要通过消息的唯一标识(MessageId)和幂等性处理
- Python AI教程之二十一:监督学习之支持向量机(SVM)算法
潜洋
人工智能Python中级支持向量机算法机器学习python
支持向量机(SVM)算法支持向量机(SVM)是一种功能强大的机器学习算法,广泛用于线性和非线性分类以及回归和异常值检测任务。SVM具有很强的适应性,适用于各种应用,例如文本分类、图像分类、垃圾邮件检测、笔迹识别、基因表达分析、人脸检测和异常检测。SVM特别有效,因为它们专注于寻找目标特征中不同类别之间的最大分离超平面,从而使其对二分类和多分类都具有鲁棒性。在本大纲中,我们将探讨支持向量机(SVM)
- 集团企业IT技术架构规划方案(基于TOGAF企业架构框架方法论)
公众号:优享智库
数字化转型数据治理主数据数据仓库架构微服务云原生
集团企业IT技术架构规划方案(基于TOGAF企业架构框架方法论)集团企业IT技术架构规划方案(基于TOGAF企业架构框架方法论)引言项目背景与目标TOGAF方法论简介规划方案概述企业现状分析与评估业务流程梳理现有IT架构评估存在问题及挑战分析架构设计原则与策略制定架构设计原则确定技术选型及标准化策略安全性、可靠性和可扩展性考虑业务架构规划与设计业务需求梳理与整合业务功能模块划分业务流程优化建议数据
- 构建决策树对于流失用户进行分类
努力学习中的阿达
最近被分配到商业分析组配合商业分析师对流失掉的客户进行研究。我最先接到的任务是根据客服部门记录的客户的流失原因,对于这些客户的流失原因做分类。商业分析师给我提供了23个类别,要求我把客户都分到这些类中。最开始我企图通过建立关键词规则,比如包含某些单词或者不包含某些单词,但是实际上发现分类的结果很差,规则首先不完备,并且彼此还可能冲突,分类的结果当然就很差。于是我就想到可以利用文本挖掘的方法,对于客
- TOGAF中的企业架构:让业务架构与数据、应用、技术架构形成闭环的魔法之旅
火山说数
数字化企业架构架构微服务云原生
前言你是否曾经有过这样一种感觉:企业在进行数字化转型时,架构之间常常感觉像是一盘散沙?业务部门、IT部门、数据分析师各自为政,技术团队则像一群“救火队员”随时准备扑灭各种系统bug。好消息是,TOGAF(TheOpenGroupArchitectureFramework)可以帮助企业打破这种局面,让业务架构(BusinessArchitecture)和其他“三A”架构——数据架构(DataArch
- 企业级应用的历史、现状与未来:技术转移与问题优化
自由鬼
行业发展IT应用探讨微服务企业级应用软件开发
企业级应用是组织生产环境的核心,旨在保障关键需求如安全、稳定、扩展性和业务连续性。在技术发展的过程中,企业级应用的架构和实现方式经历了显著的变化。本文将探讨企业级应用的历史、现状及未来,并分析技术演进中问题转移与优化的现象。一、企业级应用的历史:传统架构的复杂性在传统架构中,企业级应用的设计主要依赖于强大的中间件和数据库。例如,IBMWAS(WebSphereApplicationServer)常
- 迭代器模式详解附有代码案例分析(包含迭代器模式的源码应用分析)
hyyyya
设计模式列表java设计模式数据结构
迭代器模式一、迭代器模式的概念和角色(一)、迭代器模式的概念(二)、迭代器模式的角色二、迭代器模式的应用场景三、迭代器模式的代码示例四、迭代器模式在源码中的应用五、迭代器模式的优缺点(一)、优点(二)、缺点六、设计模式的相关博客文章链接1、七大设计原则的简单解释(包含合成复用原则),简单理解、快速入门,具备案例代码2、工厂模式详解附有代码案例分析(简单工厂,工厂方法,抽象工厂)3、单例模式详解及代
- Godot引擎开发:物理引擎高级用法_物理引擎的最佳实践与案例分析
chenlz2007
游戏开发2godot游戏引擎javaandroid材质
物理引擎的最佳实践与案例分析在上一节中,我们探讨了Godot引擎中物理引擎的基本原理和使用方法。了解了如何创建物理体、应用力和冲量、检测碰撞等基本操作。在这一节中,我们将进一步深入探讨物理引擎的高级用法,通过最佳实践和案例分析,帮助你在动作游戏中更高效地利用物理引擎,实现更加复杂和真实的物理效果。1.物理引擎性能优化在动作游戏中,物理引擎的性能优化是至关重要的。如果物理模拟不流畅,会导致游戏体验大
- python计算nino3.4指数
Mango0919
python后端matplotlib
本文定义厄尔尼诺与拉尼娜事件的方法为美国气象中心(CPC)的定义方法,即以30年为一次气候模态,三个月滑动平均的Niño3.4指数连续五个月大于0.5℃被认为是一次厄尔尼诺事件,连续五个月小于-0.5℃则被认为是一次拉尼娜事件,Niño3.4区的范围为5°N–5°S,120°–170°W。我们分析的是海表面温度(SST),我选取的是1951-1980年的数据,全球海洋温度月数据可以从美国国家气象海
- 基于 Python 的毕设选题管理系统设计与实现
赵谨言
论文python毕业设计经验分享python
标题:基于Python的毕设选题管理系统设计与实现内容:1.摘要本文介绍了一个基于Python的毕设选题管理系统的设计与实现。该系统旨在解决传统毕设选题管理方式中存在的效率低下、信息不透明等问题。通过使用Python语言和相关技术,实现了对毕设选题的信息化管理,提高了管理效率和质量。本文详细介绍了系统的需求分析、设计思路、功能实现和测试结果。系统采用了B/S架构,实现了学生选题、教师审核、管理员管
- HQL之投影查询
归来朝歌
HQLHibernate查询语句投影查询
在HQL查询中,常常面临这样一个场景,对于多表查询,是要将一个表的对象查出来还是要只需要每个表中的几个字段,最后放在一起显示?
针对上面的场景,如果需要将一个对象查出来:
HQL语句写“from 对象”即可
Session session = HibernateUtil.openSession();
- Spring整合redis
bylijinnan
redis
pom.xml
<dependencies>
<!-- Spring Data - Redis Library -->
<dependency>
<groupId>org.springframework.data</groupId>
<artifactId>spring-data-redi
- org.hibernate.NonUniqueResultException: query did not return a unique result: 2
0624chenhong
Hibernate
参考:http://blog.csdn.net/qingfeilee/article/details/7052736
org.hibernate.NonUniqueResultException: query did not return a unique result: 2
在项目中出现了org.hiber
- android动画效果
不懂事的小屁孩
android动画
前几天弄alertdialog和popupwindow的时候,用到了android的动画效果,今天专门研究了一下关于android的动画效果,列出来,方便以后使用。
Android 平台提供了两类动画。 一类是Tween动画,就是对场景里的对象不断的进行图像变化来产生动画效果(旋转、平移、放缩和渐变)。
第二类就是 Frame动画,即顺序的播放事先做好的图像,与gif图片原理类似。
- js delete 删除机理以及它的内存泄露问题的解决方案
换个号韩国红果果
JavaScript
delete删除属性时只是解除了属性与对象的绑定,故当属性值为一个对象时,删除时会造成内存泄露 (其实还未删除)
举例:
var person={name:{firstname:'bob'}}
var p=person.name
delete person.name
p.firstname -->'bob'
// 依然可以访问p.firstname,存在内存泄露
- Oracle将零干预分析加入网络即服务计划
蓝儿唯美
oracle
由Oracle通信技术部门主导的演示项目并没有在本月较早前法国南斯举行的行业集团TM论坛大会中获得嘉奖。但是,Oracle通信官员解雇致力于打造一个支持零干预分配和编制功能的网络即服务(NaaS)平台,帮助企业以更灵活和更适合云的方式实现通信服务提供商(CSP)的连接产品。这个Oracle主导的项目属于TM Forum Live!活动上展示的Catalyst计划的19个项目之一。Catalyst计
- spring学习——springmvc(二)
a-john
springMVC
Spring MVC提供了非常方便的文件上传功能。
1,配置Spring支持文件上传:
DispatcherServlet本身并不知道如何处理multipart的表单数据,需要一个multipart解析器把POST请求的multipart数据中抽取出来,这样DispatcherServlet就能将其传递给我们的控制器了。为了在Spring中注册multipart解析器,需要声明一个实现了Mul
- POJ-2828-Buy Tickets
aijuans
ACM_POJ
POJ-2828-Buy Tickets
http://poj.org/problem?id=2828
线段树,逆序插入
#include<iostream>#include<cstdio>#include<cstring>#include<cstdlib>using namespace std;#define N 200010struct
- Java Ant build.xml详解
asia007
build.xml
1,什么是antant是构建工具2,什么是构建概念到处可查到,形象来说,你要把代码从某个地方拿来,编译,再拷贝到某个地方去等等操作,当然不仅与此,但是主要用来干这个3,ant的好处跨平台 --因为ant是使用java实现的,所以它跨平台使用简单--与ant的兄弟make比起来语法清晰--同样是和make相比功能强大--ant能做的事情很多,可能你用了很久,你仍然不知道它能有
- android按钮监听器的四种技术
百合不是茶
androidxml配置监听器实现接口
android开发中经常会用到各种各样的监听器,android监听器的写法与java又有不同的地方;
1,activity中使用内部类实现接口 ,创建内部类实例 使用add方法 与java类似
创建监听器的实例
myLis lis = new myLis();
使用add方法给按钮添加监听器
- 软件架构师不等同于资深程序员
bijian1013
程序员架构师架构设计
本文的作者Armel Nene是ETAPIX Global公司的首席架构师,他居住在伦敦,他参与过的开源项目包括 Apache Lucene,,Apache Nutch, Liferay 和 Pentaho等。
如今很多的公司
- TeamForge Wiki Syntax & CollabNet User Information Center
sunjing
TeamForgeHow doAttachementAnchorWiki Syntax
the CollabNet user information center http://help.collab.net/
How do I create a new Wiki page?
A CollabNet TeamForge project can have any number of Wiki pages. All Wiki pages are linked, and
- 【Redis四】Redis数据类型
bit1129
redis
概述
Redis是一个高性能的数据结构服务器,称之为数据结构服务器的原因是,它提供了丰富的数据类型以满足不同的应用场景,本文对Redis的数据类型以及对这些类型可能的操作进行总结。
Redis常用的数据类型包括string、set、list、hash以及sorted set.Redis本身是K/V系统,这里的数据类型指的是value的类型,而不是key的类型,key的类型只有一种即string
- SSH2整合-附源码
白糖_
eclipsespringtomcatHibernateGoogle
今天用eclipse终于整合出了struts2+hibernate+spring框架。
我创建的是tomcat项目,需要有tomcat插件。导入项目以后,鼠标右键选择属性,然后再找到“tomcat”项,勾选一下“Is a tomcat project”即可。具体方法见源码里的jsp图片,sql也在源码里。
补充1:项目中部分jar包不是最新版的,可能导
- [转]开源项目代码的学习方法
braveCS
学习方法
转自:
http://blog.sina.com.cn/s/blog_693458530100lk5m.html
http://www.cnblogs.com/west-link/archive/2011/06/07/2074466.html
1)阅读features。以此来搞清楚该项目有哪些特性2)思考。想想如果自己来做有这些features的项目该如何构架3)下载并安装d
- 编程之美-子数组的最大和(二维)
bylijinnan
编程之美
package beautyOfCoding;
import java.util.Arrays;
import java.util.Random;
public class MaxSubArraySum2 {
/**
* 编程之美 子数组之和的最大值(二维)
*/
private static final int ROW = 5;
private stat
- 读书笔记-3
chengxuyuancsdn
jquery笔记resultMap配置ibatis一对多配置
1、resultMap配置
2、ibatis一对多配置
3、jquery笔记
1、resultMap配置
当<select resultMap="topic_data">
<resultMap id="topic_data">必须一一对应。
(1)<resultMap class="tblTopic&q
- [物理与天文]物理学新进展
comsci
如果我们必须获得某种地球上没有的矿石,才能够进行某些能量输出装置的设计和建造,而要获得这种矿石,又必须首先进行深空探测,而要进行深空探测,又必须获得这种能量输出装置,这个矛盾的循环,会导致地球联盟在与宇宙文明建立关系的时候,陷入困境
怎么办呢?
- Oracle 11g新特性:Automatic Diagnostic Repository
daizj
oracleADR
Oracle Database 11g的FDI(Fault Diagnosability Infrastructure)是自动化诊断方面的又一增强。
FDI的一个关键组件是自动诊断库(Automatic Diagnostic Repository-ADR)。
在oracle 11g中,alert文件的信息是以xml的文件格式存在的,另外提供了普通文本格式的alert文件。
这两份log文
- 简单排序:选择排序
dieslrae
选择排序
public void selectSort(int[] array){
int select;
for(int i=0;i<array.length;i++){
select = i;
for(int k=i+1;k<array.leng
- C语言学习六指针的经典程序,互换两个数字
dcj3sjt126com
c
示例程序,swap_1和swap_2都是错误的,推理从1开始推到2,2没完成,推到3就完成了
# include <stdio.h>
void swap_1(int, int);
void swap_2(int *, int *);
void swap_3(int *, int *);
int main(void)
{
int a = 3;
int b =
- php 5.4中php-fpm 的重启、终止操作命令
dcj3sjt126com
PHP
php 5.4中php-fpm 的重启、终止操作命令:
查看php运行目录命令:which php/usr/bin/php
查看php-fpm进程数:ps aux | grep -c php-fpm
查看运行内存/usr/bin/php -i|grep mem
重启php-fpm/etc/init.d/php-fpm restart
在phpinfo()输出内容可以看到php
- 线程同步工具类
shuizhaosi888
同步工具类
同步工具类包括信号量(Semaphore)、栅栏(barrier)、闭锁(CountDownLatch)
闭锁(CountDownLatch)
public class RunMain {
public long timeTasks(int nThreads, final Runnable task) throws InterruptedException {
fin
- bleeding edge是什么意思
haojinghua
DI
不止一次,看到很多讲技术的文章里面出现过这个词语。今天终于弄懂了——通过朋友给的浏览软件,上了wiki。
我再一次感到,没有辞典能像WiKi一样,给出这样体贴人心、一清二楚的解释了。为了表达我对WiKi的喜爱,只好在此一一中英对照,给大家上次课。
In computer science, bleeding edge is a term that
- c中实现utf8和gbk的互转
jimmee
ciconvutf8&gbk编码
#include <iconv.h>
#include <stdlib.h>
#include <stdio.h>
#include <unistd.h>
#include <fcntl.h>
#include <string.h>
#include <sys/stat.h>
int code_c
- 大型分布式网站架构设计与实践
lilin530
应用服务器搜索引擎
1.大型网站软件系统的特点?
a.高并发,大流量。
b.高可用。
c.海量数据。
d.用户分布广泛,网络情况复杂。
e.安全环境恶劣。
f.需求快速变更,发布频繁。
g.渐进式发展。
2.大型网站架构演化发展历程?
a.初始阶段的网站架构。
应用程序,数据库,文件等所有的资源都在一台服务器上。
b.应用服务器和数据服务器分离。
c.使用缓存改善网站性能。
d.使用应用
- 在代码中获取Android theme中的attr属性值
OliveExcel
androidtheme
Android的Theme是由各种attr组合而成, 每个attr对应了这个属性的一个引用, 这个引用又可以是各种东西.
在某些情况下, 我们需要获取非自定义的主题下某个属性的内容 (比如拿到系统默认的配色colorAccent), 操作方式举例一则:
int defaultColor = 0xFF000000;
int[] attrsArray = { andorid.r.
- 基于Zookeeper的分布式共享锁
roadrunners
zookeeper分布式共享锁
首先,说说我们的场景,订单服务是做成集群的,当两个以上结点同时收到一个相同订单的创建指令,这时并发就产生了,系统就会重复创建订单。等等......场景。这时,分布式共享锁就闪亮登场了。
共享锁在同一个进程中是很容易实现的,但在跨进程或者在不同Server之间就不好实现了。Zookeeper就很容易实现。具体的实现原理官网和其它网站也有翻译,这里就不在赘述了。
官
- 两个容易被忽略的MySQL知识
tomcat_oracle
mysql
1、varchar(5)可以存储多少个汉字,多少个字母数字? 相信有好多人应该跟我一样,对这个已经很熟悉了,根据经验我们能很快的做出决定,比如说用varchar(200)去存储url等等,但是,即使你用了很多次也很熟悉了,也有可能对上面的问题做出错误的回答。 这个问题我查了好多资料,有的人说是可以存储5个字符,2.5个汉字(每个汉字占用两个字节的话),有的人说这个要区分版本,5.0
- zoj 3827 Information Entropy(水题)
阿尔萨斯
format
题目链接:zoj 3827 Information Entropy
题目大意:三种底,计算和。
解题思路:调用库函数就可以直接算了,不过要注意Pi = 0的时候,不过它题目里居然也讲了。。。limp→0+plogb(p)=0,因为p是logp的高阶。
#include <cstdio>
#include <cstring>
#include <cmath&

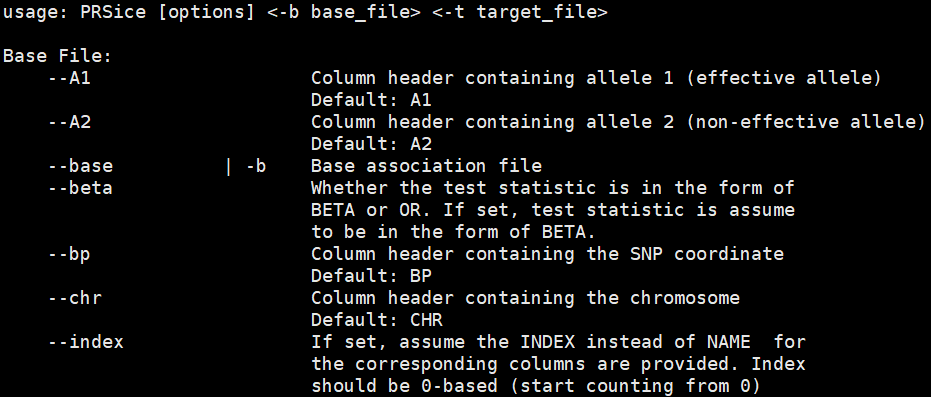
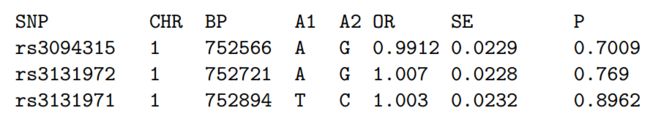 image.png
image.png
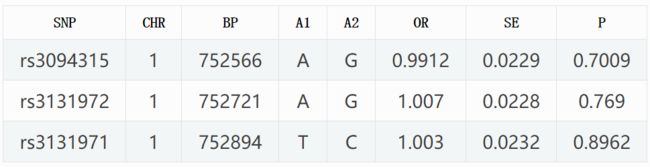 image.png
image.png
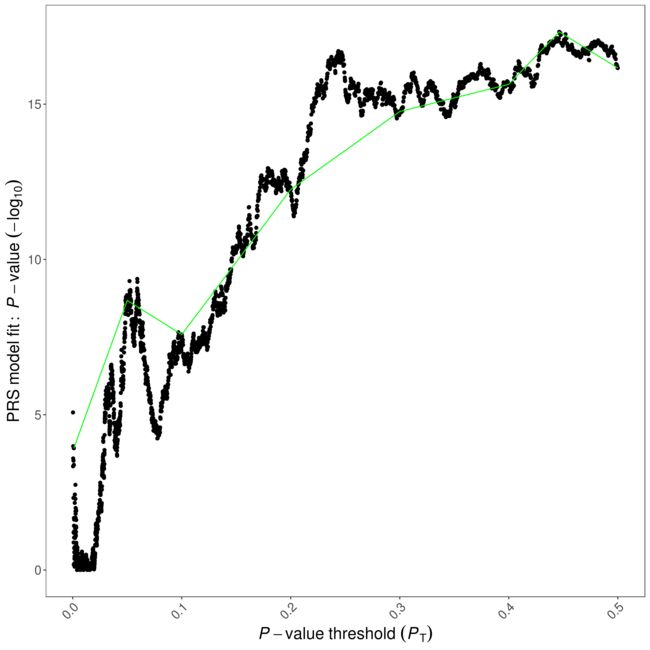 image.png
image.png image.png
image.png