原笔记地址:http://x2yline.gitee.io/microarray_data_analysis_note/
1. 芯片数据结构
芯片数据的结构如下
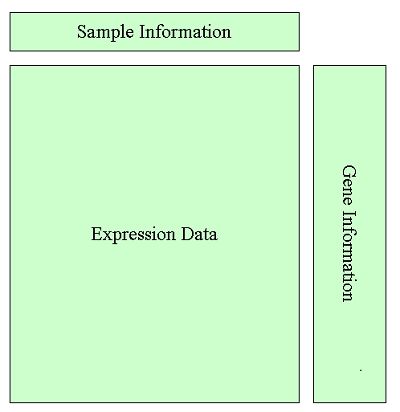
1.1 Sample Information
对于Sample Information,我们至少需要知道样本的组别,在一些更加复杂的使用设计中,我们可能还需要知道更多关于样本的信息如:
- 对于研究癌症和正常人的实验设计中,我们可能需要记录每个个体的性别,年龄等信息
- 对于动物实验很多相关的处理因素可能需要进行记录
1.2 R中的数据框
在R的数据框类型数据中,每一行代表一个实验样本,每一列代表每一个我们感兴趣的因素(包括处理因素),dataframe可以同时处理文本因子类型和数值类型的值因而非常实用
通常我们在excel中创建一个储存样本信息的表格【每一行代表一个样本,每一列代表一个因素】,并存储为csv或tab分隔的文本文件,再用R读取为dataframe【read.csv,read.delim,read.table函数】
1.3 AnnotatedDataFrame对象(样本信息)
Bioconductor的Biobase包通过一个叫做AnnotatedDataFrame对象来存储样本信息,这个对象包括:
- 每个样本的“phenotype”信息,即不同的处理或因素的信息
- 其他meta-information,如对各个处理或不同因素的描述
如我们有8个样本的芯片数据,每个芯片都有200个基因探针:
fake.data <- matrix(rnorm(8*200), ncol=8)
接下来我们需要创建AnnotatedDataFrame对象来描述这8个样本的表型:
- 包括一个数据框用来区分8个样本(即8列)的表型信息
- 一个meta数据框用了描述表型数据框中每一个因素的详细信息
## 样本的表型信息
## 注意data.frame函数会自动把字符串向量转化为因子(factor)
sample.info <- data.frame(
spl=paste('A', 1:8, sep=''),
stat=rep(c('cancer', 'healthy'), each=4))
## 对上一个dataframe列名的详细描述
meta.info <- data.frame (labelDescription =
c('Sample Name', 'Cancer Status'))
## 创建AnnotatedDataFrame对象
library(Biobase)
pheno <- new("AnnotatedDataFrame",
data = sample.info,
varMetadata = meta.info)
可以使用pData查看AnnotatedDataFrame对象的样本表型信息
pheno
## An object of class 'AnnotatedDataFrame'
## rowNames: 1 2 ... 8 (8 total)
## varLabels: spl stat
## varMetadata: labelDescription
pData(pheno)
## spl stat
## 1 A1 cancer
## 2 A2 cancer
## 3 A3 cancer
## 4 A4 cancer
## 5 A5 healthy
## 6 A6 healthy
## 7 A7 healthy
## 8 A8 healthy
1.4 ExpressionSet对象(表达矩阵与样本信息的整合)
在bioconductor中,ExpressionSet对象可以把表达矩阵和样本信息整合在一起
my.experiments <- new("ExpressionSet",
exprs=fake.data,
phenoData=pheno)
可以直接查看ExpressionSet的一些基本信息(基因数,样本数等)
my.experiments
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 200 features, 8 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: 1 2 ... 8 (8 total)
## varLabels: spl stat
## varMetadata: labelDescription
## featureData: none
## experimentData: use 'experimentData(object)'
## Annotation:
注意:如果使用上述方法创建一个
ExpressionSet对象,则一定要保证表达矩阵的列与样本信息的行相对应,软件没有办法对这些信息进行检查
1.5 基因信息
我们创建好了一个ExpressionSet对象,但是这个对象却没有对基因的注释及相关信息,bioconductor的一个瑕疵就是它允许用户把表达数据与基因信息完全分离开(但大多数人对此都欣然接受)
1.6 ExpressionSet对象的其他可选信息
处理表达数据(exprs)和样本信息(phenoData)外ExpressoinSet对象还允许添加其他的一些可选信息
- annotation:基因注释信息的环境(这里有点不太理解???)
- se.exprs:表达矩阵中每个元素对应的标准差,因此该数据也是一个矩阵
- notes:描述该实验相关信息的字符串
- description:
MIAMEclass的一个对象,即Minimun Information About a Microarray Experiment
1.7 内置示例数据及简单分析
对于Affymetrix公司的芯片数据,bioconductor提供了一个特殊的ExpressionSet数据类型,即AffyBatch,affydata包提供了一个示例数据Dilution,Dilution即为一个AffyBatch对象
# BiocInstaller::biocLite("affydata")
library(affydata)
data(Dilution)
Dilution
## AffyBatch object
## size of arrays=640x640 features (30414 kb)
## cdf=HG_U95Av2 (12625 affyids)
## number of samples=4
## number of genes=12625
## annotation=hgu95av2
## notes=
phenoData(Dilution)
## An object of class 'AnnotatedDataFrame'
## sampleNames: 20A 20B 10A 10B
## varLabels: liver sn19 scanner
## varMetadata: labelDescription
pData(Dilution)
## liver sn19 scanner
## 20A 20 0 1
## 20B 20 0 2
## 10A 10 0 1
## 10B 10 0 2
注:如果是第一次查看Dilution这个数据,bioconductor会自动联网并更新基因的注释信息,这可能会花一段时间。
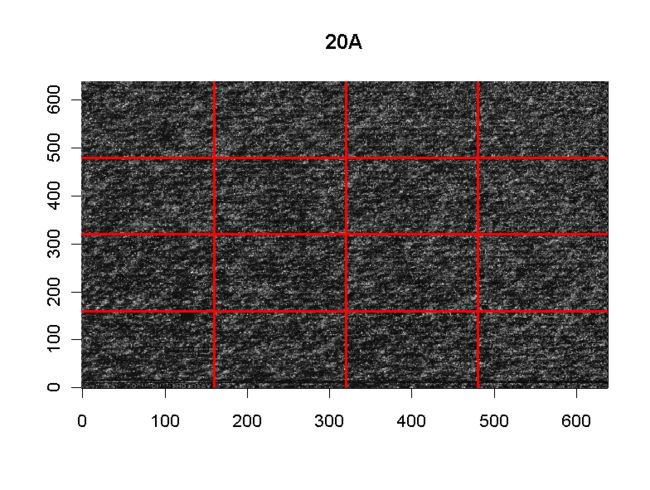
首选我们可以看一下芯片的扫描图像,如果出现了异常亮区或异常暗区,则说明质量可能存在问题(被刮了一下)如下图
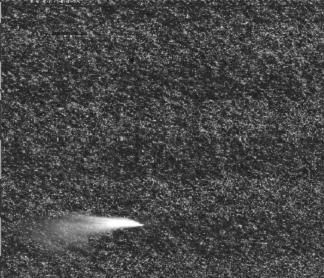
par(mfrow=c(2, 2))
image(Dilution)
par(mfrow=c(1, 1))
也可以用箱式图及密度分布图查看每个芯片的表达量分布(注意表达量自动进行log2处理),如果出现了某个芯片与其他芯片的差异很大,说明该芯片质量可能存在问题
boxplot(Dilution, col=1:4)
hist(Dilution, col=1:4, lty=1)

affy包也提供提取特定探针数据的函数
geneNames(Dilution)[1:3]
## [1] "100_g_at" "1000_at" "1001_at"
probeNames(Dilution)[1:48]
## [1] "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at"
## [9] "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at" "100_g_at"
## [17] "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at"
## [25] "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at" "1000_at"
## [33] "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at"
## [41] "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at" "1001_at"
可看出一个序列(一个基因或RNA对应一个probset)对应16个探针(通常一个probset对应11~20种探针,每种探针会重复数百次)
# probset函数截取特定探针的数据
# 产生一个Probset对象组成的list
# probset对象包含了表达数据
probeset(Dilution, c("100_g_at", "34803_at"))
## $`100_g_at`
## ProbeSet object:
## id=100_g_at
## pm= 16 probes x 4 chips
##
## $`34803_at`
## ProbeSet object:
## id=34803_at
## pm= 16 probes x 4 chips
##
ps <- probeset(Dilution, c("100_g_at", "34803_at"))[[2]]
# perfect match
pm(ps)
## 20A 20B 10A 10B
## 235143 435.3 267.0 315.0 192.3
## 121393 206.0 143.8 155.0 118.0
## 99468 349.8 302.8 277.0 171.8
## 196621 567.0 399.0 480.0 322.3
## 256831 160.5 140.5 137.3 97.8
## 351719 474.0 312.8 401.0 240.0
## 361833 561.5 365.5 468.8 319.0
## 111309 742.8 503.0 622.5 346.0
## 407736 713.5 440.0 503.0 303.0
## 407737 736.0 468.0 564.5 327.0
## 238835 808.3 527.5 516.0 321.0
## 173838 463.3 322.3 372.0 228.5
## 320847 240.0 177.0 172.0 136.8
## 195435 129.0 80.0 92.0 75.3
## 327084 193.0 128.3 150.0 96.3
## 12704 120.0 98.0 109.0 80.5
# mis-match
mm(ps)
## 20A 20B 10A 10B
## 235783 230.0 140.3 180.0 111.0
## 122033 109.5 78.0 94.5 67.8
## 100108 162.0 133.8 119.0 85.0
## 197261 223.0 132.3 174.8 102.5
## 257471 164.0 104.0 130.5 76.3
## 352359 371.0 267.0 301.0 187.5
## 362473 487.0 275.5 352.3 200.0
## 111949 439.0 286.0 311.5 183.0
## 408376 393.0 240.3 282.3 154.0
## 408377 655.8 415.8 453.8 282.0
## 239475 885.0 548.3 511.0 338.0
## 174478 215.8 137.0 145.0 100.0
## 321487 134.3 96.0 102.0 88.0
## 196075 113.0 67.3 107.0 62.3
## 327724 132.3 69.3 91.5 67.0
## 13344 107.0 137.0 95.3 64.0
对该基因的探针数据(未经过log2处理)进行可视化:
plot(c(1,16), c(50, 900), type='n',
xlab='Probe', ylab='Intensity')
for (i in 1:4) lines(pm(ps)[,i], col=i)
for (i in 1:4) lines(mm(ps)[,i], col=i, lty=2)

## pm-mm的值进行可视化
plot(c(1,16), c(-80, 350), type='n',
xlab='Probe Pair', ylab='PM - MM')
temp <- pm(ps) - mm(ps)
for (i in 1:4) lines(temp[,i], col=i)
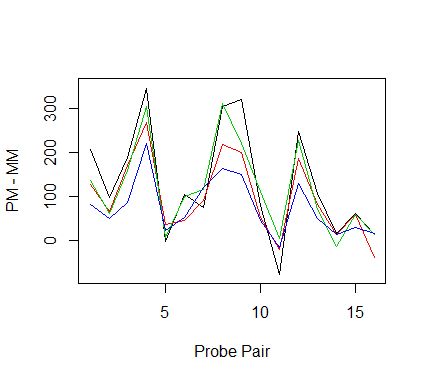
同时affy包也能查看RNA的降解情况,因为RNA的降解都是从5`向3`开始的,这16(通常每个probset即每个RNA对应这11~20个不同位置的探针)个探针的靶序列也是从RNA的5`向3`方向逐渐靠近的
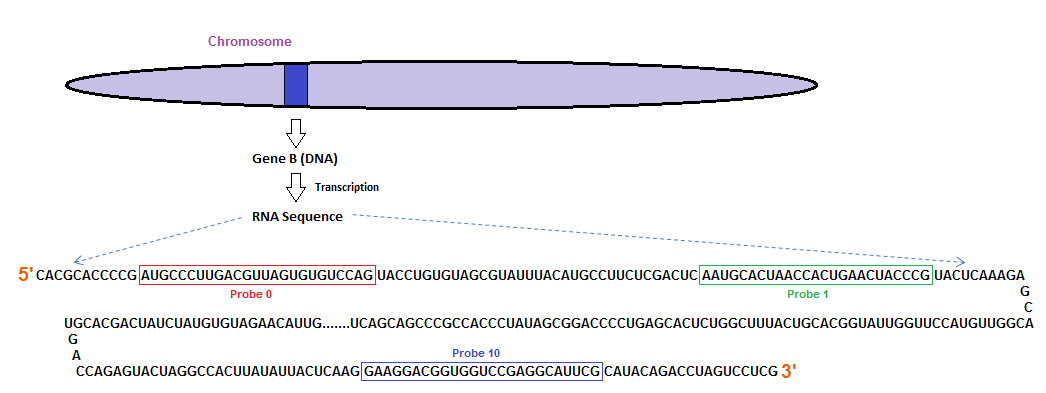
以杂交的信号为纵坐标,探针的位置作为横坐标作图(其中纵坐标进行了缩放和移动以便对不同样本的斜率进行比较),可以推测5`端的探针序列对应的RNA相对与3`来说密度较低,信号也越低,因此曲线整体呈上升趋势
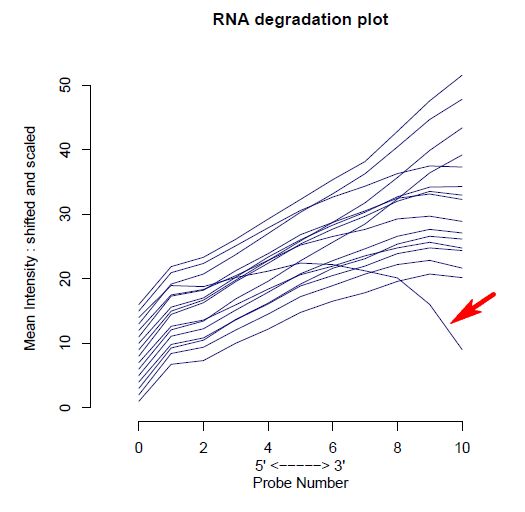
比斜率更重要的是芯片之间的比较,理想状况下各芯片的曲线是平行的,而降解得很多的样品其5' 到 3' 线段的斜率会比其他样品大,该数据显示每个位置的探针降解速度都不太一致(斜率呈分段变化),但样品之间的降解一致[1][2],通常从5‘到3’端的第5个探针认为是比较稳定的
degrade <- AffyRNAdeg(Dilution)
plotAffyRNAdeg(degrade, col=1:4)

下图为存在异常降解芯片脱颖而出的数据:
2. 使用bioconductor读取Affymetrix芯片数据
affy包提供了ReadAffy函数使创建AffyBatch对像变得简单,该函数还提供了GUI(graphical user interface)界面方便用户的使用,然而我们需要对分析过程进行文档化,为了分析的可重复性我们应该避免使用GUI界面
由CEL文件创建AffyBatch对象
library(affy)
# There is a function list.celfiles that
# makes it easy to get a list of the CEL files
fns <- list.celfiles("data/CEL_file_dir",
full.names = TRUE)
fns[1:2]
## [1] "CEL_file_dir/CL2001011101AA.CEL" "CEL_file_dir/CL2001011102AA.CEL"
# read CEL data
my.data <- ReadAffy(filenames=fns)
构建样本信息的数据AnnotatedDataFrame对象
其中样本的信息文件(data_file_list.txt)格式如下:
scan.name sample_name type
CL2001011101AA ALL_1 A
CL2001011104AA ALL_2 A
CL2001011105AA ALL_3 A
使用函数Biobase包的read.AnnotatedDataFrame读取注释文件,设置第二列为行名:
adf <- read.AnnotatedDataFrame(file.path("data", "/data_file_list.txt"),
header = TRUE, sep = "\t", row.names = 2)
adf
## An object of class 'AnnotatedDataFrame'
## rowNames: ALL_1 ALL_2 ... AML_28 (72 total)
## varLabels: scan.name type
## varMetadata: labelDescription
# 对列名的注释信息(可选)
varMetadata(adf) <- data.frame(labelDescription=
c("file name", "disease classification"))
phenoData(my.data) <- adf
也可以再构建一个description,即MIAME对象
miame <- new("MIAME", name = "someone",
lab = "your lab name",
title = "Gene expression data ...",
pubMedIds = c("your pubmed ID"))
description(my.data) <- miame
description(my.data)
## Experiment data
## Experimenter name: someone
## Laboratory: your lab name
## Contact information:
## Title: Gene expression data ...
## URL:
## PMIDs: your pubmed ID
## No abstract available.
另一种方法为之间使用read.affybatch函数之间构建AffyBatch对象
abatch <- read.affybatch(filenames = fns,
phenoData = adf,
description = miame)
3. 数据预处理
bioconductor把low-level processing(即把探针级别【一个基因对应多个探针】的数据转为为基因级别的数据,从而使一个probset对应一个表达量值)分为了4个步骤,每个步骤都可以选择不同的算法,这些方法的不同极有可能会导致后面high-level analyses的差异
这四个步骤为:
- Background correction
- Normalization (on the level of features = probes)
- PM-correction(包括只使用PM,丢弃掉MM,或者把PM-MM作为校正的PM等)
- Summarization(把一个probset中的多个probe对应的多值总结为1个值)
3.1 背景信号的校正
背景校正的目的是为了消除人为的或芯片本身存在的噪声所带来的误差
bgcorrect.methods()
## [1] "bg.correct" "mas" "none" "rma"
- mas:使用 MAS 5.0 算法
- none:不做校正
- rma:使用 RMA 算法
MAS 5.0 算法
MAS 5.0 的背景校正方法为把每个芯片(每个CEL文件)分为16个区,在每个区中最最暗的2%作为每个区的背景水平,在对该芯片的每个探针用这16个背景值的加权平均进行校正,每个区的权重取决于该探针与芯片中这16个区域中心的距离。

RMA算法
RMA算法只对PM进行背景校正,MM的值不做校正(放弃MM,因为几乎有1/3的MM值是大于对应的PM的),该模型的假设为PM的值是由两个部分组成的即S=X+Y,其中S为实际检查到的信号,X代表有探针杂交产生的信号服从指数分布Exp(α),Y代表背景信号服从正态分布N(μ, δ^2),Y只取大于0的部分,并且X与Y是独立的

最后校正过后的X值=E(X|S = s),其中s为该探针的信号强度,E(X|S = s)表示在检测到的信号强度为s的情况下X的期望值,具体推导可以看大牛的博士论文:http://bmbolstad.com/Dissertation/Bolstad_2004_Dissertation.pdf,暂时没有时间来研究,先搁在这里。
还有一个gcrma的算法,考虑到不同序列gc含量与序列亲和力的关系进而估计不同背景信号大小,需要安装gcrma包bgc.gcrma = gcrma(Data),该命令包括了背景校正,标准化和summarization这几个步骤。
不同算法的结果
library(affydata)
data(Dilution)
d.mas <- bg.correct(Dilution[, 1], "mas")
d.rma <- bg.correct(Dilution[, 1], "rma")
bg.with.mas <- pm(Dilution[, 1]) - pm(d.mas)
bg.with.rma <- pm(Dilution[, 1]) - pm(d.rma)
par(mfrow=c(1,2))
hist(bg.with.mas)
hist(bg.with.rma)
par(mfrow=c(1,1))
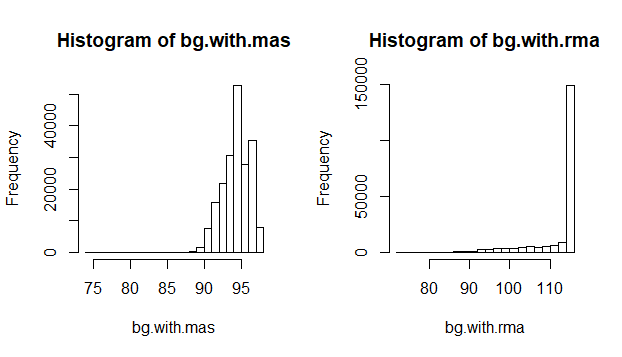
summary(data.frame(bg.with.mas, bg.with.rma))
## X20A X20A.1
## Min. :74.53 Min. : 72.4
## 1st Qu.:93.14 1st Qu.:113.7
## Median :94.35 Median :114.9
## Mean :94.27 Mean :112.1
## 3rd Qu.:95.80 3rd Qu.:114.9
## Max. :97.67 Max. :114.9
在该示例数据中,RMA算法估计的背景值比MAS 5.0估计的背景值大,而且RMA算法的背景值比较恒定
# 两种算法的差值分布
hist(bg.with.rma - bg.with.mas)
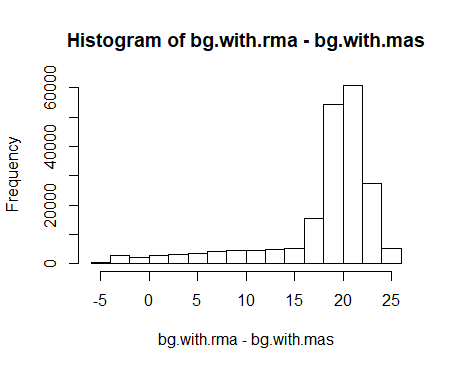
差值大部分为20,可以由芯片的信号值分布看一看20究竟有多大
tmp <- data.frame(pm(Dilution[, 1]), mm(Dilution[, 1]))
colnames(tmp) <- c("PM", "MM")
summary(tmp)
## PM MM
## Min. : 76.0 Min. : 77.3
## 1st Qu.: 137.0 1st Qu.: 120.3
## Median : 225.0 Median : 164.5
## Mean : 507.3 Mean : 323.5
## 3rd Qu.: 489.0 3rd Qu.: 313.0
## Max. :23356.3 Max. :17565.3
boxplot(tmp, ylim=c(0, 1000))

可以看出20越占大部分信号值的1/10,不同算法的差值还是比较大的
3.2 summarization=Quantication
暂时先不管normalization和PM correction这一步,summarization的目的就是把所有同一probset(即同一基因mRNA)的PM和MM探针对转化为一个数值,这个数值是对目标基因表达量的最佳估计,其方法有:
express.summary.stat.methods()
## [1] "avgdiff" "liwong" "mas" "medianpolish" "playerout"
使用不同的summarization方法对probeset进行定量,expresso函数把所有预处理步骤都整合在了一起
tempfun <- function(method) {
expresso(Dilution, bg.correct = FALSE,
normalize = FALSE, pmcorrect.method = "pmonly",
summary.method = method)
}
ad <- tempfun("avgdiff")
al <- tempfun("liwong")
am <- tempfun("mas")
ar <- tempfun("medianpolish")
MA plot
MA plot即M-versus-A plot,在芯片数据处理出现之前也称为Bland-Altman plot,是由发明者名字命名的,而MA plot是对M与A作图而得名,M是minus的缩写,代表两个值之差,A是add的缩写,代表两个值之和。有研究者也把MA plot称为Ratio-Intensity (RI) plots。
MA plot的作用是为了展示两个值几乎处处相等的变量(x和y)之间的关系,为了展示两个变量之间的变化关系,大多数人的思维都是把x与y分别作为横轴和纵轴进行绘图,如果y=x,则该图呈45度角的直线(如下图中左边图的蓝色直线),可以通过查看点形成的直线偏离预期直线的多少来衡量系统偏差,然而该图存在以下几个缺点:
- 人的视觉对水平线比更敏感
- 不同坐标轴的刻度可能会使预期参考直线偏离45度
- 很难从直观上衡量偏离一条线性的大小
MA plot的处理方法是把该直线顺时针旋转45度,把参考对角线变为直线,具体做法是把(x+y)/2作为横轴,(y-x)作为纵轴,则参考直线变为一条水平线,如下方右图,这样可以很清楚的在视觉上展示两个相等的变量之间偏离参考值的大小,即存在的系统误差的大小
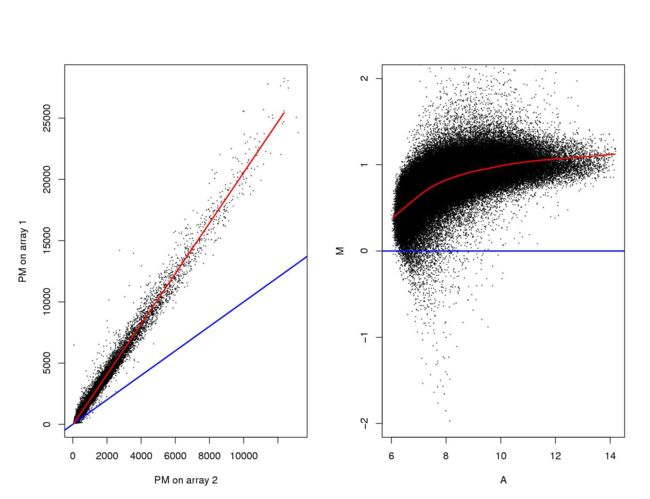
bioconductor中的MA plot
affy包的函数mva.pairs可以快速地做出MA plot(也可以使用MAplot函数),接下来我们使用MA plot来比较不同summarize方法之间的差异,mva.pairs函数的输入参数就是各变量形成的一个dataframe
## ar进行了log2处理
temp <- data.frame(exprs(ad)[, 1],
exprs(al)[,1],
exprs(am)[, 1],
2^exprs(ar)[, 1])
dimnames(temp)[[2]] <- c("Mas4", "dChip",
"Mas5", "RMA")
affy::mva.pairs(temp)
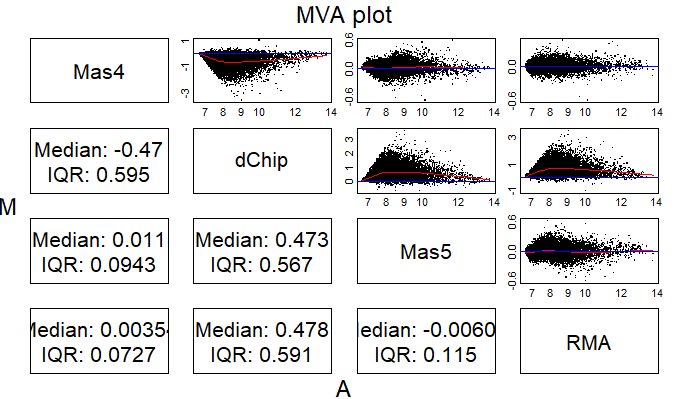
上图中的差异也有可能是由于我们没有在summarization之前进行校正,接下来我们使用RMA方法进行背景校正,使用百分位数标准化方法,并且只用PM值进行数据定量汇总
tempfun <- function(method) {
expresso(Dilution,
bgcorrect.method = "rma",
normalize.method = "quantiles",
pmcorrect.method = "pmonly",
summary.method = method)
}
ad <- tempfun("avgdiff")
al <- tempfun("liwong")
am <- tempfun("mas")
ar <- tempfun("medianpolish")
## ar进行了log2处理
temp <- data.frame(exprs(ad)[, 1],
exprs(al)[,1],
exprs(am)[, 1],
2^exprs(ar)[, 1])
dimnames(temp)[[2]] <- c("Mas4", "dChip",
"Mas5", "RMA")
mva.pairs(temp)
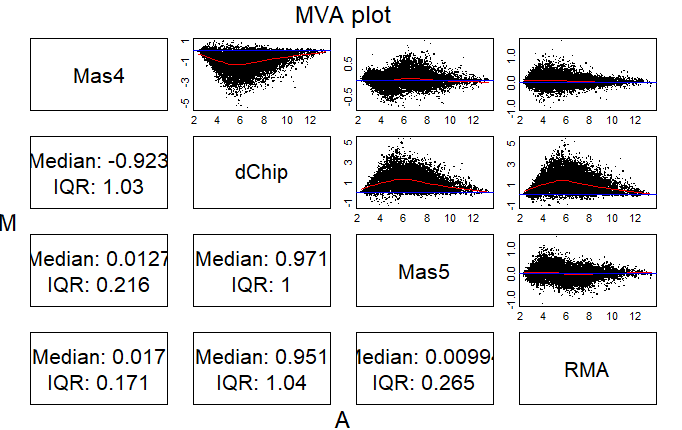
summarization的详细过程
每个算法都包括了四个步骤的不同处理方式(即一个算法包含4个小算法),如MAS 4.0 包括MAS,constant,subtractmm,avgdiff
不同算法的组合如下表(摘抄自:NAR文献Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons):
| Background correction | Normalization | PM-correction | Summarization | Common name |
|---|---|---|---|---|
| mas | invariantset | mas | liwong | |
| none | quantiles | mas | liwong | |
| mas | quantiles | mas | liwong | |
| none | invariantset | mas | liwong | |
| mas | quantiles | pmonly | liwong | |
| none | constant | mas | liwong | |
| rma | quantiles | pmonly | liwong | |
| mas | constant | mas | liwong | |
| rma | invariantset | pmonly | liwong | |
| none | quantiles | pmonly | liwong | |
| mas | invariantset | pmonly | liwong | |
| none | constant | pmonly | liwong | |
| none | invariantset | pmonly | liwong | Li–Wong |
| rma | constant | pmonly | liwong | |
| mas | constant | pmonly | liwong | |
| gcrma | invariantset | pmonly | liwong | |
| gcrma | constant | pmonly | mas | |
| gcrma | quantiles | pmonly | mas | |
| gcrma | invariantset | pmonly | mas | |
| none | invariantset | mas | mas | |
| none | quantiles | mas | mas | |
| gcrma | quantiles | pmonly | liwong | |
| none | constant | mas | mas | |
| mas | invariantset | mas | mas | |
| mas | quantiles | mas | mas | |
| mas | constant | mas | mas | mas5.0 |
| none | constant | mas | medianpolish | |
| gcrma | quantiles | pmonly | medianpolish | gcrma |
| gcrma | invariantset | pmonly | medianpolish | |
| mas | constant | mas | medianpolish | |
| gcrma | constant | pmonly | medianpolish | |
| none | quantiles | mas | medianpolish | |
| gcrma | constant | pmonly | liwong | |
| mas | quantiles | mas | medianpolish | |
| none | invariantset | mas | medianpolish | |
| mas | invariantset | mas | medianpolish | |
| none | invariantset | pmonly | mas | |
| none | quantiles | pmonly | mas | |
| none | constant | pmonly | mas | |
| mas | constant | pmonly | mas | |
| mas | quantiles | pmonly | mas | |
| mas | invariantset | pmonly | mas | |
| rma | quantiles | pmonly | mas | |
| rma | constant | pmonly | mas | |
| mas | constant | pmonly | medianpolish | |
| rma | invariantset | pmonly | mas | |
| mas | quantiles | pmonly | medianpolish | |
| rma | constant | pmonly | medianpolish | |
| rma | quantiles | pmonly | medianpolish | rma |
| rma | invariantset | pmonly | medianpolish | |
| mas | invariantset | pmonly | medianpolish | |
| none | quantiles | pmonly | medianpolish | |
| none | invariantset | pmonly | medianpolish | |
| none | constant | pmonly | medianpolish |
MAS 4.0 算法(MicroArray Suite 4.0)
MAS4.0代表一个完整的数据处理算法,对背景校正,标准化,PM校正,probeSet定量4个完整步骤都有其特定的计算过程:
- 背景校正算法也叫做MAS,即使用2%的信号作为背景如前所述
- 标准化算法为constant,即把所有芯片的中位数缩放致一个相等的常数
- PM校正为PM-MM,即subtractmm
- summarization使用叫做avgdiff的算法,即把PM-MM的离群值(偏离均值大于3倍标准差)删除后计算平均值为基因的表达量值
对avgdiff的评价:
- 对所有的探针都一视同仁(没有加权重)
- 对PM-MM的值只是简单的相加
- 可能会把一些有趣的探针给删除掉(离群值)
- 可能会产生负值(大约有1/3的MM大于PM)
- 不能有效利用其他芯片的数据(没有建立一个模型)
MAS 5.0 算法(MicroArray Suite 5.0)
mas5.0的4个步骤中,除了标准化为constant外,其余均名为mas
PM校正方法名为mas:即把PM-CT作为校正的PM,CT为change threshold是一个调整后的MM,以保证CT值小于PM值,CT值算法为
- 如果MM小于PM则CT=MM
- 如果MM大于PM,则CT校正为一个比PM较小的数,如下图所示

summarization方法也称为mas:即Signal = anti-log of Tukeybiweight(log (PM –CT))
点评:mas 仍然没有用到不同芯片的数据即建立一个模型
开发了mas 5.0后,affymetrix公司不再投入大量精力开发算法,但提供了一套可以检验算法的数据The Affy Latin Square Experiment.用这个数据可以证明mas 5.0比mas 4.0(avgdiff)表现更优越
dChip(一个芯片数据分析软件,哈佛)算法也较Li-Wong算法
Li-Wong算法【由现北大研究员李程与现斯坦福教授王永雄开发】包括三个小部分,不包括背景校正过程,标准化为invariantset(以后再了解),PM校正方法为只使用PM值或PP和MM值都使用(可自定义),summarization为一个统计学模型称为Li-Wong算法,所有芯片的数据都得到了很好的利用,但是如果芯片数目比较少时可信度较差
[图片上传失败...(image-b9fc36-1525698429039)]
[图片上传失败...(image-b1b7e2-1525698429039)]
其模型最终PM-MM值为探针亲和力$\varphi_{j}$与表达量$\theta_{i}$的乘积,i代表样本,j代表探针,$v_{i}$代表非特异性杂交的基线水平,文章地址为:http://www.pnas.org/content/98/1/31
RMA(Robust Multichip Analysis)算法
RMA算法是2003年由Rafael A. Irizarry(Bekerley)等人提出的,前面已经大致介绍了背景校正的方法,标准化使用的是一种叫做median polish的方法(以后再介绍),该方法完全丢弃了MM值(他们给出的理由是因为太多MM信号值大于PM值的情况,因此考虑MM的值会引入很大的变异)【他们的想法或许是正确的】
像dChip一样,该算法的summarization方法是建立在一个统计学模型之上的:
[图片上传失败...(image-a12c9f-1525698429039)]
其中芯片编号为i,j代表探针
模型的参数由多个芯片的数据进行拟合得到,与dChip不同的是$\epsilon$的值是经过对数处理的,这充分考虑到了值越大的探针其变异也越大的特点
PDNN模型(考虑杂交的热力学因素)
以上算法都是从数学层面上进行处理,而没有试图解释不同探针之间的信号差异来源,以及是什么决定了非特异性结合。通常这些都与探针的序列,及杂交的热力学性质相关
Li Zhang提出了Position-Dependent Nearest Neighbor(PDNN) 模型(Nat Biotech, 2003; 21:818).不像dChip和RMA,PDNN模型参数的估计只需要单个芯片的数据(很大程度上是由于参数数目比较少)。该模型考虑序列对杂交的影响,把信号强度值与序列联系起来。
该模型的参数有:
- 碱基对在序列中的位置k
- 与临近碱基对的作用,除了知道k,还需要知道k-1和k+1
该模型的实现需要单独安装一个包affypdnn,在载入这个包之后,会自动载入相关函数,并更新可选的模型
# registering new summary method 'pdnn'.
# registering new pmcorrect method 'pdnn' and 'pdnnpredict'.
express.summary.stat.methods()
## [1] "avgdiff" "liwong" "mas" "medianpolish" "playerout" "pdnn"
需要注意的是PDNN模型没有按照标准的expresso4个步骤处理数据,该模型直接从百分位数校正开始,可以只使用PM或PM和MM同时进行拟合,背景的估计整合到了能量和表达参数里面。
需要使用expressopdnn这个expresso的变体实现PDNN模型的运算。
4. 质量控制(QC)
4.1 简单质控
即前面的简单分析部分
第一步是看图形,有没有异常的区域
接着可以看看表达值的分布情况,看有没有异常的芯片
image(Dilution)
boxplot(Dilution, col = 2:5)
hist(Dilution, col = 2:5, lty = 1)
4.2 simpleaffy包进行质控
simpleaffy质控得到的结果都是按照MAS5.0算法产生的,如校正因子,背景信号强度等都是MAS 5.0算法的结果
require(simpleaffy)
Dil.qc <- qc(Dilution)
首先我们可以看看每个芯片的背景值是否有可比性,差别非常大的异常值
# 各芯片背景均值
sort(avbg(Dil.qc))
## 10B 20B 10A 20A
## 54.25830 63.63855 80.09436 94.25323
# 各芯片背景最大值
sort(maxbg(Dil.qc))
## 10B 20B 10A 20A
## 57.62283 68.18998 83.24646 97.66280
# 各芯片背景最小值(MAS 5.0算法)
sort(minbg(Dil.qc))
## 10B 20B 10A 20A
## 49.22574 60.01397 77.32196 89.52555
芯片标准化时,标准的Affymetrix流程为乘上一个校正因子(scaling factors)使各芯片的中位数相同,Affymetrix要求可比较的芯片之间的校正因子不能超过3
# 标准校正流程中的校正因子
sfs(Dil.qc)
## [1] 0.8934013 1.2653627 1.1448430 1.8454067
max(sfs(Dil.qc))/min(sfs(Dil.qc))
## [1] 2.065597
接下来是percent of present值(即有多少基因按照Detection calls算法是确实存在表达且被检测到的)是否有可比性,如果有离群值或低于30,高于60的值说明可能存在质量问题
percent.present(Dil.qc)
## 20A.present 20B.present 10A.present 10B.present
## 48.79208 49.82178 49.37822 49.75842
Affymetrix设计了一些参考管家基因(通常是GAPDH或beta-actin)的3‘,middle和5’的探针,其中actin的3‘/5'的值应该是小于3,GAPDH的3'/5'值小于1(5’端可能存在部分降解,可与RNA降解图结合起来看),否则可能存在问题
ratios(Dil.qc)
## actin3/actin5 actin3/actinM gapdh3/gapdh5 gapdh3/gapdhM
## 20A 0.6961423 0.1273385 0.4429746 -0.06024147
## 20B 0.7208418 0.1796231 0.3529890 -0.01366293
## 10A 0.8712069 0.2112914 0.4326566 0.42375270
## 10B 0.9313709 0.2725534 0.5726650 0.11258237
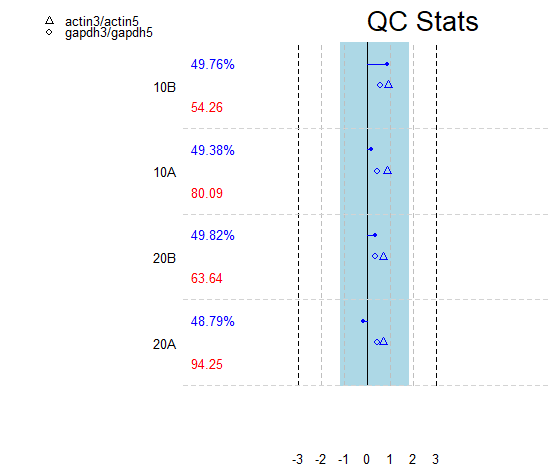
也可以之间对qc结果进行作图查看
中间的黑色实线代表fold change为0的参考线,黑色虚线代表上调或下调基因的参考线即fold change绝对值超过3
蓝色区域代表各样本的scale factor理论上的范围跨度(其确定方式为以各芯片的scale factor均值为中心先两侧各延伸1.5个fold change单位,即跨度刚好为3)
横线可是蓝色或红色(蓝色代表芯片的scale factor是可接受的,红色代表有芯片的scale factor超过了理论范围不具有可比性,下图为蓝色说明各芯片的scale factor比较相似具有可比性);
actin的3‘/5'的值应该是小于3,GAPDH的3'/5'值小于1,任何超过该范围的标记为红色否则为蓝色;
左边的数值上面的是percent of present(差异超过10%则标记为红色),下面的是average background(差异超过20个单位则标记为红色,但是这个值可能需要调整)
plot(Dil.qc)
4.3 RNA降解图使用affy包查看,可参考之前的部分
4.4 依赖统计学模型的质控包affyPLM
affyPLM是一个与RMA模型相似的probe-level model (PLM)从而进行质控
library("affyPLM")
pset <- fitPLM(Dilution)
模型拟合的残差可以用image函数来可视化显示,可以很清楚地判断是否有异常残差值的出现,网址http://www.plmimagegallery.bmbolstad.com/收集了很多特征性的图片,而Dilution这个数据集基本正常,通常该图片没有瑕疵的情况很少,小的瑕疵一般都可以接受,通过RMA这个稳健的算法基本可以把误差消除掉,而当污点比较大时,我们需要再看其他的质控数据决定是否应该舍弃该芯片。
par(mfrow = c(2, 2))
# 第三张芯片的原始图片
image(Dilution[, 3])
# 第三张芯片的校正后值图片(默认为type=“weight",值越大颜色越淡)
image(pset, type = "weights", which = 3)
# 第三张芯片的残差的图片(红色渐变色表示正值越大,白色表示接近0,蓝色渐变色表示负值越大)
image(pset, type = "resids", which = 3)
# 第三张芯片的残差的图片(红色表示所有正值,白色表示接近0,蓝色表示所有负值)
image(pset, type = "sign.resids", which = 3)

下面这个就是有问题的图片,除原始图片不太清楚外,其他图片都明显显示有人为误差,即一个“环”,就需要看下一步的QC数据是否有较大异常决定去留

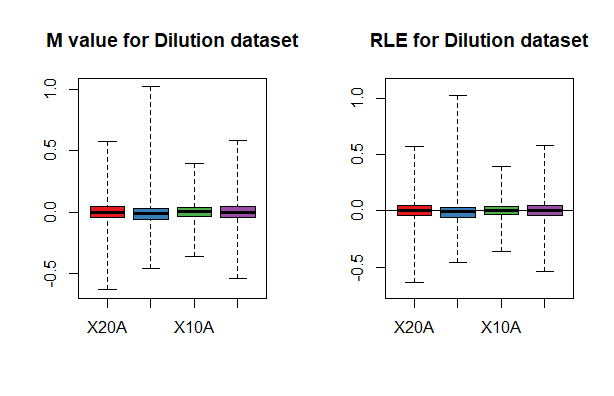
还可以用箱式图查看M值(相对于表达量为中等的那个芯片)分布也叫做Relative Log Expression的分布。 如果有芯片的中位数不是0或跨度明显大于其他芯片,则可能存在误差,这个两个图都是一模一样的,不过名称不同而已
require(RColorBrewer)
cols <- brewer.pal(8, "Set1")
par(mfrow=c(1,2))
Mbox(pset, main="M value for Dilution dataset", col=cols)
RLE(pset,
main="RLE for Dilution dataset",
ylim=c(-0.7, 1.1),
col=cols)
par(mfrow=c(1,1))
RLE(pset, type = "stats")
## 20A 20B 10A 10B
## median 0.002359773 -0.007542337 0.003051789 0.001902185
## IQR 0.092560253 0.089580148 0.076335304 0.093406833
RLE(pset, type = "values")[1:4, ]
## 20A 20B 10A 10B
## 100_g_at 0.08993939 -0.074287948 0.026305114 -0.02630511
## 1000_at 0.11551276 -0.007420133 0.007420133 -0.04293690
## 1001_at 0.08565396 -0.071191808 -0.043903380 0.04390338
## 1002_f_at 0.11275165 -0.045783998 -0.013287804 0.01328780
接下来就是看Normalized Unscaled Standard Error (NUSE)值的分布是否异常,与M值(RLE值)的思想差不多,NUSE是每个基因的标准差与该基因中所有芯片中的标准差中位数的比值,因此芯片具有可比性的条件是其所有基因的标准差几乎相同,即NUSE的中位数为1,且分布大致相当。

par(mfrow=c(1,2))
boxplot(pset, main="NUSE for Dilution dataset", col=cols)
NUSE(pset, main="NUSE for Dilution dataset",
col=cols,
ylim=c(0.88, 1.23))
par(mfrow=c(1,1))

我们再来看一个异常的RLE和NUSE图,其中第二个芯片与image一样都偏离了正常值

本文为MD Anderson芯片分析课程的lecture 7笔记,最后的异常值图片参考子bioconductor书籍Bioinformatics and Computational Biology Solutions Using R and Bioconductor
其他参考:
http://repository.countway.harvard.edu/xmlui/bitstream/handle/10473/4713/hummel_affy_normalization.pdf
http://www.math.usu.edu/~jrstevens/stat5570/Bowles.pdf
-
http://arrayanalysis.org/main.html#RD ↩
-
https://www.jianshu.com/p/859a7a7d040f ↩