本文译自Challenges and Opportunities in Protease-Activated Receptor Drug Development
摘要
蛋白酶活化受体(PAR)是一类独特的GPCR,它接收来自胞外蛋白酶的信号。PAR在血管生成、炎症和癌症中具有重要功能,是重要的药物靶点。PAR的一个独特特征是其蛋白水解激活过程不可逆,生成不能扩散的系固配体。尽管GPCR已被证明是最成功的一类可用药靶标,但专门针对PAR的制剂的开发仍然具有挑战性。因此,研究人员采取了多种不同的方法来开发调节PAR功能的药物。在这里,我们概述了针对PAR开发的治疗药物的多样性。我们还进一步讨论了PAR偏置信号和受体区隔、翻译后修饰和二聚化的影响等药物开发的重要考量因素。
前言
GPCR介导多种生物学过程,其功能失调与多种病理状态有关,是一个很有吸引力的药物靶点。大多数GPCR是由肽、激素或离子等天然小分子激活的,这些小分子很容易被合成类似物模拟,因此,这类受体是美国食品和药物管理局(FDA)批准的用于治疗的最大靶标。蛋白酶活化受体(PAR)是GPCR大超家族中很特别的成员,它能介导细胞对胞外蛋白酶的反应,是重要的药物靶点。然而,PAR蛋白水解活化的性质导致了不可逆的活化,这与大多数其他GPCR不同。PAR激活的这种不可逆性,对受体特异性拮抗剂的开发提出了不寻常的挑战。
自1991年发现PAR1以来,人们对PAR1的体内功能、其他PAR的体内功能、不同细胞环境中的受体信号传导特性以及受体信号传导和功能的调控方面的研究都取得了重大进展。然而,针对特定PAR选择性的变构配体药物的研究一直困难重重。因此,多种不同的方法已被用于开发针对PAR的药物。在这篇综述中,我们讨论了针对PARs的治疗药物的多样性及其临床疗效。我们还将进一步讨论靶向PAR的药物研发的机遇和挑战,包括偏置信号受体属性的新视角以及会影响受体信号反应和药物开发的转译后修饰和二聚作用等等。
PAR功能和激活
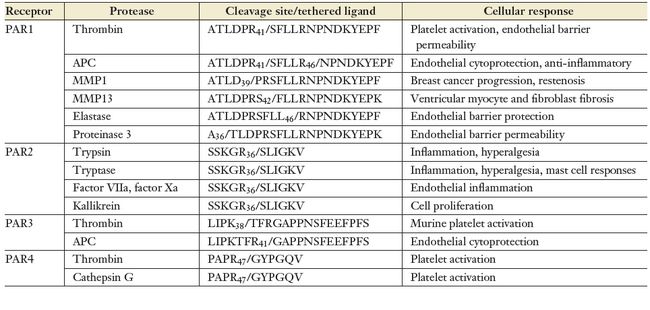
PAR在血管生理、发育、炎症和癌症进展等方面具有重要作用。哺乳动物基因组可以编码四个PAR。PAR1,PAR家族原型,传递细胞对凝血酶的反应,后者是凝血级联的关键效应蛋白酶。凝血酶也能激活PAR3和4,而PAR2是胰蛋白酶样丝氨酸蛋白酶激活的。PAR1是在寻找人类血小板中凝血酶应答受体时被发现的第一个PAR。PAR2是接下来在基因组文库筛选时发现的,它能够介导胰蛋白酶应答。PAR3和4依次被发现,它们介导小鼠血小板中凝血酶信号传导,如此,不同PAR在不同细胞中差异化表达,但是作用方式类似。PAR1、PAR3和PAR4主要在血管系统的各类型细胞中表达,包括血小板、成纤维细胞、内皮细胞和平滑肌细胞,它们是体内凝血酶信号的主要效应因子。然而,其他蛋白酶可以以类似凝血酶的方式或水解N末端特定位点来激活这些受体。PAR2在血管、肠和肺细胞中表达,主要介导与组织损伤相关的炎症反应。与其他亚型类似,PAR2是由多种蛋白酶(包括胰蛋白酶和上游凝血因子)激活的。因此,作为PAR激活的生理调节因子的特定蛋白酶取决于组织和细胞类型。
PAR的激活起始于胞外N端结构域被水解,从而产生一个新的可以与受体结合的N端,进而跨膜传递信号。模拟受配体序列的合成肽可以在不通过凝血酶或者蛋白酶水解的方式激活PAR1。凝血酶可以识别特定的位于PAR1、PAR3和PAR4 N端的水解位点。第二个相互作用发生在凝血酶的阴离子外结合位点和PAR1、PAR3N端结构域的酸性hirudin样序列之间,这个作用提高了特异性凝血酶结合和水解的精准程度。PAR4没有这个第二作用位点,与凝血酶的亲和力低了不少。与其他PAR类似,trypsin-like蛋白酶识别并水解PAR2的N端位点,暴露配体结合位点,进而激活PAR2。水解后,被激活的PAR跨膜螺旋发生构象变化,促进了与异三聚体G蛋白的相互作用。PAR1识别很多配体,可以与包括Gαi,Gαq,Gα12/13在内的许多G蛋白相互作用。PAR2和PAR4似乎也可以与Gαq和Gα12/13相互作用,而PAR3至少在某些细胞类型中没有表现出这些作用,。除了异三聚体G蛋白,激活的PAR还可以通过与β- arrestin和转化生长因子β-激活激酶结合蛋白1 (TAB1:MAPK激活的支架)相互作用。最近的研究表明,PAR显示出激活偏好性。偏好活性或功能选择性是指不同配体稳定GPCR独特活性构象的能力,这种活性构象可促进不同信号反应的激活。几项研究已经清楚地证明不同蛋白酶在特定位点水解PAR,会导致独特的连接肽配体的生成,从而促进不同异三聚体G蛋白亚型或β-arrestins信号的传导。这些发现提高了选择性激活或阻断PAR激活引起的某些信号通路的配体开发的可能性,可能成为重要的治疗手段。
PAR1药物研发的多样性
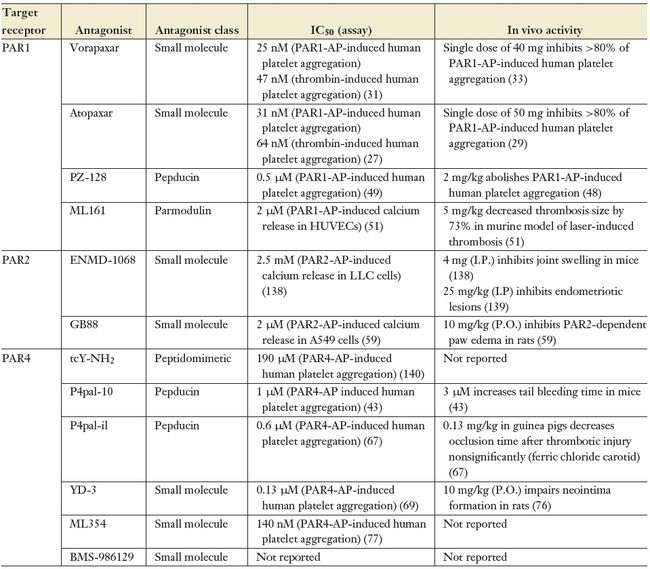
目前唯一针对PAR的临床药物是PAR1拮抗剂vorapaxar。早期研究观察到PAR1主要介导凝血酶诱导的血小板功能,随后二十年来科学家们积极开发PAR1的小分子拮抗剂,并最终在2014年成功研发出预防血栓性心血管疾病的vorapaxar,并获得FDA批准。由于广泛的药物开发项目,PAR1拮抗剂的质量和数量远远超过了针对PAR家族其他成员的拮抗剂。事实上,关于PAR1拮抗的方法有相当多的报道。最早的PAR1抑制剂是直接针对受体上凝血酶结合外泌体或凝血酶水解位点区域的功能阻断抗体。尽管这种方法在体内外都有效,但有一个主要限制是:当激动剂浓度增加时,抑制活性被轻松克服,这表明需要改善PAR1功能阻断抗体的KD值。类似地,一系列肽和拟肽PAR1抑制剂被用于PAR1功能的早期研究。这些药物一方面模拟受体的内源性结合配体序列,另一方面可以阻止随后受体激活的修饰。该方法贡献了几个关键实验试剂,包括一系列NH2 -酰基四肽,如BMS-197525和BMS-200261,以及RWJ系列吲哚和吲唑衍生物,如RWJ-56110和RWJ-58259。虽然它们在早期研究中广泛应用于PAR1功能研究,但这些肽类拮抗剂通常缺乏特异性、效用或两者都缺,而且许多具有部分激动剂的功能,因此目前的应用非常有限。在这里,我们重点概述三种不同类型的PAR1拮抗剂——小分子拮抗剂、pepducins和parmodulin——它们与受体的不同位点结合,应用于最近的研究中。
PAR1小分子拮抗剂
一些小分子PAR1拮抗剂已经被开发出来,其中,阿托帕沙(atopaxar)和沃拉帕沙(vorapaxar)已经在大规模的临床试验中得到检验。atopaxar和vorapaxar都能够与PAR1第二个细胞外环内的配体结合位点或附近可逆结合,并与内源性激活配体竞争,发挥经典竞争拮抗剂的作用。
Atopaxar,原名E5555,是一种分子量为609 Da的双环脒衍生物。Atopaxar抑制一种氚标记合成激动肽 [3H]SFLLRN与PAR1的结合,并抑制人血小板对凝血酶和一种PAR1特异性激动肽的凝集反应。在体内,atopaxar在几个血栓形成的小动物模型中显示出抗血栓形成的活性,包括光化学诱导的豚鼠血栓形成。在这个模型中,Atopaxar将血栓血管阻塞的时间延长了大约2倍,而没有显著影响皮肤出血时间。鉴于Atopaxar在体内的疗效的效用,该药物被用于对急性冠脉综合征(LANCELOT-ACS)和慢性冠脉疾病(LANCELOT-CAD)患者的人体试验。然而,在II期安全试验中,服用高剂量Atopaxar的患者肝酶升高,QT间期延长导致快速心律失常,并且大出血事件发生率增加,显示出明显的靶上和脱靶副作用。因此,Atopaxar没有进展到III期试验。
Vorapaxar,原名SCH530348,也是一种可逆的、竞争性的小分子PAR1拮抗剂。Vorapaxar是一种合成的三环3-苯基吡啶类似物,其分子量为591 Da。Vorapaxar具有抑制人类凝血酶引起的血小板聚集和PAR1激活的功能。最初在老鼠和猴子中进行的vorapaxar药代动力学分析证明Vorapaxar有很高的口服生物利用度和13 h的半衰期。在人体中,vorapaxar耐受性好,持续时间长,终端血浆半衰期为126-269小时,单次给药生物利用度大于90%。与atopaxarII期临床试验结果相反,长期使用沃拉帕沙并没有导致肝功能异常。Vorapaxar已在两项大型III期临床试验中得到评估:用于急性冠脉综合征临床事件减少(TRACER)的凝血酶受体拮抗剂试验和用于动脉粥样硬化血栓缺血性事件二级预防(TRA2P-TIMI 50)的凝血酶受体拮抗剂试验。虽然TRACER未能达到其主要终点(一个复合心血管死亡,心肌梗死,中风,复发性缺血,或紧急冠状动脉血管重建),TRA 2P-TIMI 50试验显示出使用vorapaxar的明显好处。具体来说,vorapaxar与标准护理抗血小板治疗联用时,心血管事件发生率从10.5%降至9.3%,P < 0.001,且无明显致命性出血效应。因此,vorapaxar于2014年年中被FDA批准用于预防心肌梗死的血栓并发症或外周动脉疾病。
除了这两种临床试验药物外,其他几种小分子PAR1拮抗剂仍在实验研究中使用。最常用的是SCH79797和SCH203009,它们在多种细胞类型中显示出对par1介导事件的有效抑制,并且在体内有效。然而,这些药物的疗效和选择性都不如atopaxar和vorapaxar,而且有脱靶效应。
因此,临床检测的两种药物atopaxar和vorapaxar已成为PAR1拮抗剂实验研究的金标准。尽管在小分子的发展中取得了成功,仍有一系列其他抑制PAR1功能的方法,如下所述。
靶向PAR1的Pepducins
基于Pepducin的PAR1抑制剂有一种抑制剂已进入初步临床,但更多仍在研究细节。Pepducins是一类独特的GPCR拮抗剂,其作用机制是阻断受体与异三聚体G蛋白的相互作用。PAR1是在最初概念验证研究中被检验的第一个GPCR。Pepducins包含一个肽序列,该肽序列对应介导G蛋白相互作用的目标GPCR的胞内环(ICLs)或C末端的一个区域。这一短肽序列N端耦联了棕榈酸酯(pal),可以锚定于细胞膜,从而阻止G蛋白与目标相互作用位点的结合。因此,Pepducins似乎通过竞争性地结合和隔离G蛋白发挥作用,从而阻止了GPCR激活引起的下游信号传导。这种作用机制暗示了一个有争议的观点,即单个GPCR通过独特的相互作用决定因素与G蛋白结合。然而,尽管存在特异性方面的问题,抗PAR1 pepducin PZ-128(以前是P1pal-7)最近在一项有限的I期研究中对冠心病患者进行了评估。PZ-128通过模拟PAR1的ICL3作用于胞内PAR1- G蛋白界面,并已被证明可以选择性地抑制PAR1介导的人血小板活化。在豚鼠和狒狒动物模型中,PZ-128能快速有效地损伤动脉血栓形成,且对出血没有任何影响。在第一个人体研究中,PZ-128被应用于具有临床症状或冠状动脉疾病危险因素的31位患者,并显示出剂量依赖的选择性抑制PAR1介导的血小板激活,且不影响血小板激活其他受体激动剂,如PAR4肽受体激动剂、二磷酸腺苷、胶原蛋白。PZ-128的血浆半衰期为1.3-1.8小时,这些作用在24小时内是可逆的。有趣的是,在注射后6 - 24小时,观察到在一定剂量下抑制活性的后期峰值,推测是由于高亲脂性pepducin的重新分配。此外,在最高剂量时出现了明显的急性过敏反应。尽管如此,大约600名接受非紧急经皮冠状动脉介入治疗的患者被列入PZ-128进行II期安全性研究的计划。
Parmodulins:靶向PAR1的小分子拮抗剂
另一类不同的PAR1拮抗剂是最近报道的parmodulins,这是一类利用复杂而又有偏好性的G蛋白偶联PAR1模式的小分子。与pepducins类似,parmodulins与PAR1的胞内表面结合,并干扰活化的受体- G蛋白偶联,而不是在胞外干扰凝血酶水解或配体结合。在最初的研究中,筛选了大约30万个小分子来抑制血小板颗粒的分泌。这些研究发现,具有1,3-二氨基苯核的化合物可以选择性地抑制PAR1介导的血小板致密颗粒的分泌。对先导化合物ML161的修饰获得了一个详细的构效关系模型。这些化合物以细胞内的PAR1为目标,而不改变受体的胞外配体结合位点。
值得注意的是,PAR1信号是通过Gαq而不是通过Gα13通路被封闭的,体外和体内都是如此。对这种差异性的一个关键解释是,受体-G蛋白耦联可能特异性调节PAR1的凝血和促炎效应,而这种效应主要是由Gαq介导的。事实上,在不阻断保护性激活蛋白C (APC)介导的PAR1信号通路或诱导内皮损伤的情况下,parmodulins蛋白也可以抑制血栓前PAR1信号通路。这些观察结果与原位PAR1拮抗剂(如atopaxar、vorapaxar和其他小分子)对所有PAR1介导的信号事件都有抑制作用形成直接对比。这些发现提出了一种诱人的可能性,即功能选择性受体拮抗剂可以阻止病理性的但不阻止细胞保护性的PAR1介导的细胞信号事件。然而,关于受体选择性的问题仍然存在:第一代parmodulins似乎抑制了包括PAR4在内的几种GPCRs,这些受体在螺旋8中具有共性,这意味着需要进一步的研究来充分理解这类PAR1拮抗剂的特异性和作用机制。
PAR2和PAR4的药理学以及它们的拮抗剂
除了PAR1外,PAR2和PAR4也已成为重要的药物靶点,而PAR3迄今还未成为任何主要药物研发的靶点。类似于PAR1的研究,开发有效阻断PAR2或PAR4不可逆蛋白水解激活的药物也面临着巨大的困难。因此,基于PAR1所采取的不同策略被用于开发能够抑制PAR2或PAR4功能的药物。这促进了对PAR2和PAR4阻断剂研究的发展,这些阻断剂包括一些有希望的小分子抑制剂在体内外都表现出药效,不过还没有进展到临床。
针对PAR2的药物研发
PAR2是PAR家族中第二个被发现的成员,是一个胰蛋白酶激活的受体。它在多种组织中广泛表达,并与炎症、神经发育和癌症相关。因此,阻断PAR2活性可能提供治疗益处。然而,在一定条件下激活PAR2也是有益的。一些使用动物模型的研究高度暗示了PAR2在关节炎、炎症性肠病(包括结肠炎和辐射引起的肠炎症)和过敏原引起的哮喘中的重要作用。类似于PAR1的研究,已经采取了多种策略来扰乱PAR2的功能,包括使用功能阻断抗体、小分子抑制剂和pepducins。最初针对PAR2开发的小分子拮抗剂是基于胰蛋白酶水解产生的结合配体序列的肽药物。在体内实验中ENMD-1068抑制了胰蛋白酶诱导的PAR2活化,降低了关节炎症;然而,由于缺乏疗效,它未能进入临床试验。GB88是一种较新的、强效的、可逆的拮抗剂,可在体内外阻断内源性蛋白酶和合成肽激动剂对PAR2的激活;在大鼠结肠炎模型中,它已被证明可以减轻炎症。有趣的是,GB88仅选择性地阻断某些PAR2刺激的信号通路,包括Ca2+动员,但不阻断MAP激酶激活。针对PAR2结合配体序列的PAR2功能阻断抗体可以在体外抑制胰蛋白酶介导的水解,在体内减少PAR2介导的关节炎症,但尚未进展到临床试验。PAR2靶向的pepducins已被开发并显示出部分激动剂和拮抗剂活性。PAR2 pepducin P2pal-18S在体内表现出强大的拮抗活性,可以抑制炎症反应。然而,与PAR2的ICL3同源的PAR2 pepducin P2pal-21表现出部分激动剂活性。因此,与PAR1的情况类似,在体内PAR2 pepducins作为PAR2介导炎症的治疗效用需要进一步研究。
PAR4针对PAR4的药物研发
与靶向PAR1和PAR2的药物相比,PAR4药理抑制剂的发展明显缓慢,这在很大程度上是由于对PAR4生理功能的了解有限。事实上,PAR4最典型的作用是在血小板中,长期以来,它被认为在血栓素诱导的血小板激活中起主要的冗余作用,这抑制了PAR4抑制剂开发的热情。然而,在vorapaxar临床试验中观察到的PAR1抑制的局限性,以及最近发现的血小板之外的PAR4功能,重新激起了人们对开发PAR4拮抗剂的兴趣,并在最近做出了一些有希望的努力。
PAR4功能阻碍抗体
第一个PAR4拮抗剂是一个兔多克隆抗PAR4功能阻断抗体。该抗体靶向人PAR4凝血酶水解位点的肽序列。该抗体抑制了转染的Rat1细胞中凝血酶水解PAR4,中断了小鼠肺成纤维细胞中PAR4介导的钙信号通路,并在伴随PAR1抑制的情况下破坏了血栓素诱导的人血小板聚集——尽管使用浓度非常高。尽管这是一种成功的方法,但直到最近,阻断功能的抗par4抗体才被用于实验。最近开发的几种功能阻断型抗PAR4抗体包括针对PAR4凝血酶切割位点的类似兔多克隆抗体和针对PAR4 N端负离子区的类似抗体,以及针对受体这两个区域的一系列小鼠单克隆抗体。然而,一些抗PAR4抗体表现出令人惊讶的特异性缺乏,它们不仅抑制凝血酶诱导的人血小板聚集,还抑制作用于PAR4、P2Y12或GPVI受体的激动剂诱导的血小板聚集。其他PAR4抗体似乎疗效有限,最近开发的一系列单克隆抗PAR4抗体在PAR4的阴离子区或凝血酶水解位点或附近结合,仅在细胞表达系统中部分地阻止凝血酶水解和PAR4活化。相比之下,最近针对PAR4凝血酶切割位点的多克隆抗体,在类似的基于血小板的功能测定和体外人血栓模型中,具有高度特异性和惊人的有效性。然而,使用基于功能阻断抗体的方法靶向PAR4是否对未来的实验和潜在的临床研究有用,仍有待确定。
针对PAR4的Peptidomimetics
与早期的PAR1拮抗剂一样,基于PAR4的结合配体序列的肽和肽类似物同样被用作PAR4初始拮抗剂。该方法的关键实验试剂为(反式肉桂基)-YPGKF-NH2 (tcY-NH2)。这个经过修饰的PAR4激活肽可以结合到但不能激活PAR4,从而组织了PAR4又到的鼠血小板的高浓度聚集。tcYNH2还能够抑制凝血酶诱导的人类血小板聚集,尽管表明tcY-NH2是人类PAR4有效抑制剂的证据有限。此外,有报道称tcY-NH2在PAR2突变体PAR2 F240S中起激动剂的作用,这引起了人们对其作为治疗剂潜在发展的严重担忧。
靶向PAR4的Pepducins
抗PAR4的Pepducins与抗PAR1Pepducins同时被研发。在最初的研究中,抗PAR4 pepducin P4pal-10 (N-pal-SGRRYGHALR-NH2)抑制了高达85%的由凝血酶引起的人和小鼠血小板聚集。P4pal-10的体内评估结果显示,在小鼠尾巴出血时间测定中,血小板依赖性止血效果显著。然而,与PAR1 pepducins一样,P4pal-10和其他抗PAR4 pepducins的特异性也存在争议。例如,P4pal-10显示对PAR1、GPVI和血栓素TP受体诱导的血小板活化的活性。事实上,P4pal-10一直作为全Gαq信号抑制剂用于最近的研究。为了避免广泛的Gαq抑制,人们开发出一种不同的抗PAR4 pepducin,它模拟PAR4的ICL1 (N-pal-ATGAPRLPST-NH2 (P4-pal-i1),前面讲述的报道于PAR1的ICL3。在靶向一个不同的受体的胞内区域时,P4-pal-i1似乎通过不同的机制发挥作用,该机制包括破坏PAR1- PAR4异二聚体、损伤PAR1-和PAR4介导的细胞事件。虽然作为凝血酶介导的细胞效应的抑制剂明显有效,但抗PAR4 pepducins缺乏特异性仍然是一个重要的问题,使用这种方法的实验研究应该谨慎解释。
针对PAR4的小分子拮抗剂
虽然已经开发了几种小分子PAR4拮抗剂,但这些研究少有发表。吲哚唑衍生物YD-3是这类化合物中研究最多的。在相对低的浓度下,YD-3抑制凝血酶激活PAR4诱导的人血小板聚集,部分抑制人血小板聚集。这种作用似乎是特异性的,因为还没有报道称该化合物对其他血小板激动剂诱导的聚集有影响。YD-3已经在一些研究中用于检测PAR4在血小板功能、血管生成和炎症中的功能。然而,由于其高亲脂性,YD-3的体内效用有限,目前正在努力优化该母化合物的整体溶解度和药效学特性。ML354是最近发现的分子量为282 Da的替代吲哚,对PAR4的选择性较好。它可以有效地抑制PAR4诱导的血小板反应。最后,BMS-986120是一系列咪唑噻二唑和咪唑吡嗪衍生物的先导化合物,它们可抑制血小板凝血酶诱导的人血小板聚集,并在I期临床试验中对预防、治疗或同时预防、治疗血栓栓塞性疾病的安全性和耐受性进行了评估(https://clinicaltrials. gov/ct2/show/NCT02208882)
PAR多态性的生物学意义
尽管研究人员已经报道了许多PAR单核苷酸多态性(SNPs),但只有少数已被证明影响PAR功能,并对药物开发有意义。有趣的是,在PAR1中发现了三个潜在的重要多态性,它们都位于基因调控区域。外显子2起始位点上游14个核苷酸处的插入序列发现了一个腺苷酸(A)-胸腺苷酸(T)转位。这个A - T转位变异影响PAR1的密度和功能,并已被证明影响心血管疾病的预后和癌症的预后。具体而言,AA基因型增加ST段抬高型心肌梗死患者发生缺血性事件的风险,并与肾癌转移风险增加相关。第二种多态性发生在启动子调控区(rs11267092)的-506处,通过13碱基重复序列(5_-CGGCCGCGGGAAG-3_)的插入/删除而产生。该13碱基插入/删除变体也与一些临床结果有关,其插入的纯合性降低了静脉血栓栓塞的风险。有趣的是,缺失等位基因与乳腺癌、胃癌和食道癌的预后改善有关。最后,翻译起始位点上游(-1426 C/T; rsXYZYXZYXZ)的C/T突变与复发性妊娠失败(可能与胎盘中PAR1水平较低有关)和早产有关,这表明PAR1功能在妊娠结局中扮演了一个有趣的角色。
与PAR1变异相比,PAR2中已知的变异尚未与显著的临床结果关联。相反,PAR2的变异似乎会影响受体的药理学。例如,PAR2中发现的240F > S多态性涉及受体第二个胞外loop区域,而S等位基因导致对蛋白水解和非蛋白水解受体激活的敏感性显著降低。第二种PAR2变异涉及一个621C > T多态性(rs631465),它似乎影响mRNA二级结构并增加稳定性,继而导致PAR2水平升高,这与一组韩国儿童患特应性疾病风险增加的报道一致,提示PAR2受体水平在炎症反应的调节中起作用。
PAR家族中最有趣的受体变异来自于PAR4,最近的证据表明PAR4中的SNP在不同的患者群体中调节受体的表达和功能。PAR4的120A > T SNP (rs773902)尤其值得注意,120T基因型与PAR4表达增加和受体敏感性相关,这可能与当前抗血小板药物的耐药性和较差的心血管结局相关。PAR4基因120T变异的频率非常高(在某些人群中>80%),并且在种族上具有双态性,在一组北美154人的族群中,有63%的黑人是该突变型,而白人的比例仅有19%。值得注意的是,来自人类基因组多样性项目的数据显示,SNP rs773902并不是区域特异性的,在撒哈拉以南非洲抽样的50-80%的人,以及大约三分之二的巴布亚人和美拉尼西亚人为PAR4 120T。值得注意的是,药理研究表明,小分子拮抗剂YD-3对PAR4诱导的血小板激活的强原位拮抗作用仅表现在120A基因型的病人,对基因型120T的患者几乎没有作用,这表明这种变体可能为针对PAR4的药物开发产生重大影响。
偏好PAR的激动剂与药物研发
GPCRs是具有多种不同构象状态的动态分子。因此,不同的受体激动剂可以稳定独特的同一GPCR的活性构象并促进激活不同的信号效应分子,如异三聚体G蛋白质或β-arrestins。PAR通过N末端的蛋白酶水解被激活,这暴露了一个供配体结合的序列,配体和受体发生分子内结合,从而触发跨膜信号。这是不是意味着不同的PAR活化蛋白酶会在同一位点使之水解,从而触发类似的信号级联,并表现出线性功效?然而,事实并非如此,PAR1就是最好的例证。
在体外培养的人类内皮细胞中,凝血酶在精氨酸41位点水解激活PAR1,促进其耦合Gα12/13 Gαq蛋白质。随后,凝血酶水解的PAR1介导小GTPase RhoA和其他信号效应因子的激活,从而促进粘附连接的解体和肌动蛋白细胞骨架的重组,导致内皮屏障的破坏(炎症反应的标志)。相反,当抗凝蛋白酶APC与其辅因子内皮蛋白C受体结合时,会水解PAR1上不同的位点——R46——并通过激活Rac1而不是RhoA信号来诱导内皮屏障的稳定。与凝血酶不同,APC激活PAR1后不触发耦合Gα12/13或Gαq信号,而是通过β-arrestins信号优先促进内皮屏障维持。代替APC激活PAR1的一个合成肽代表验证了这些反应。此外,APC水解和PAR1介导细胞保护反应需要PAR1定位于小泡微域中,但凝血酶激活PAR1就不需要。总的来说,这些研究是最早证明内源性GPCR在天然激动剂激活时有差异表现的研究之一。
APC是一种天然存在于人血浆中的抗凝血和抗炎介质。人重组APC,也被称为Drotrecogin alfa,被FDA批准用于严重脓毒症的治疗。严重脓毒症是一种由不受控制的炎症和弥散性血管内凝血引起的危及生命的临床疾病。然而,人重组APC由于疗效不足和出血增加而退出临床使用。除了严重脓毒症外,APC在多种器官损伤的临床前模型中也被证明具有保护作用。APC的细胞保护作用包括抗炎、抗凋亡和稳定内皮屏障。最近的研究表明,APC突变体选择性地表达细胞保护活性而不表达抗凝血活性,可降低由菌血症或脓毒症模型引起的死亡率。此外,在临床前研究中,缺乏抗凝活性和维持与野生型APC相似信号传导特性的APC突变体减少了缺血性卒中引起的脑损伤,增强了对缺血/再灌注损伤的心脏保护。这些APC的研究清楚地说明了利用PAR偏置激动剂进行药物开发的机会和潜力。基质金属蛋白酶(MMPs)也被证明在不同的位点水解和激活PAR1。人们首先在乳腺癌中发现MMP1在适当位置水解PAR1,进而引起Ca2+动员和乳腺癌细胞迁移和侵袭。MMP1在血管内皮细胞、血小板、成纤维细胞和巨噬细胞中表达。MMP1从D39位水解激活PAR1,进而触发Gα12/13介导的RhoA信号、MAP激酶激活、血小板形状变化,但不引起强烈的Ca2 +动员或血小板聚集,这一点与凝血酶不同。一种模拟MMP1产生PAR1系固配体的肽激动剂产生了类似的效应。凝血酶和MMP1在动脉损伤后血管平滑肌细胞和动脉狭窄中也诱导了不同的反应。除了MMP1, MMP13还可以在心室肌细胞中一个独特的F43位点上水解和激活PAR1,并可能促进偏置信号传导,但这仍有待充分研究。炎症性中性粒细胞分泌弹性蛋白酶和proteinase-3,它们分别在不同的位点——L45和A36——水解并激活PAR1。与凝血酶激活PAR1不同,弹性蛋白酶和proteinase-3 激活PAR1后信号优先通过Gαi促进MAP激酶激活。模拟弹性蛋白酶或proteinase-3生产栓系配体序列的合成肽,在表达PAR1的细胞模型中表现出相似的表型。然而,弹性蛋白酶或proteinase-3在何种生理环境下选择性激活PAR1以促进偏置信号仍有待确定。
PAR2和PAR3也可以被多种细胞外蛋白酶水解和激活,似乎可以促进偏置信号反应。然而,在考虑药物开发潜力之前,还需要进一步的研究来更好地理解蛋白酶特异性偏置信号的选择性、受体特异性信号特性和生理学意义。此外,许多蛋白酶,如基质金属蛋白酶和中性粒细胞蛋白酶,针对各种各样的蛋白质,包括其他的PAR以及血管系统和其他组织中与PAR无关的细胞功能。因此,利用特定的蛋白酶选择性激活特定的PAR或利用特定的蛋白酶选择性抑制剂抑制蛋白酶介导的PAR1激活的策略可能具有挑战性。
PARs的翻译后修饰
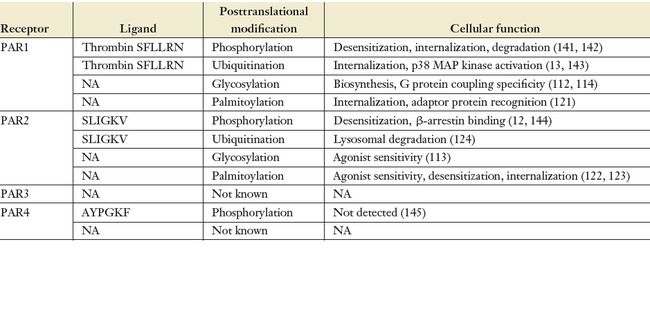
翻译后修饰是GPCRs功能的一部分。除了磷酸化外,大多数(如果不是所有的话)A类GPCRs都被天冬酰胺(N)糖基化修饰。大多数(约90%)A类GPCRs的N末端包含N-糖基化N- x - s /T共识位点,而约30%受体的胞外环内包含共识位点。然而,共识位点的范围和充分利用可能随着给定的GPCR而变化,在正常和病理条件下也可能不同。考虑到糖基化作用发生在GPCRs的胞外结构域,这些位点对配体识别也很重要,因此糖基化作用可以影响GPCR -配体相互作用也就不足为奇了,因此糖基化作用是药物发现的重要考虑因素。
PAR1包含5个N链糖基化的共识位点,3个位于N端,2个位于胞外loop2,所有这些位点似乎都有糖基化修饰。PAR2 N端和胞外loop2中各有一个N糖基化位点。PAR4仅在N端有一个N糖基化共识位点,而PAR3有两个,一个在N端,另一个位于胞外loop3。GPCRs N糖基化能够帮助初生蛋白在翻译和向细胞表面转移过程中正确折叠,与此功能一致,PAR1的N端糖基化对其向细胞表面的高效运输也非常重要。然而,在胞外loop2的PAR1的糖基化起着独特的作用。最近的研究表明,PAR1胞外loop2的糖基化可以稳定thrombin诱导PAR1产生优先耦联Gα12/13的活性构象,而不是耦合Gαq蛋白,但是对其耦联Gαi或β-arrestin-1并没有影响。糖基化的异质性表明,GPCRs可能以包含不同糖基结构的受体群体的形式存在。这表明PAR1可能存在一个整体的活跃状态,使用不同的分子结构耦联不同的G蛋白亚型或β-arrestin。除除了PAR1,PAR2的N端糖基化作用会影响类胰蛋白酶,但不影响胰蛋白酶水解和激活受体,这说明糖基化作用直接影响蛋白酶的识别和受体的激活。视网膜紫质GPCR的N糖基化序列中自然发生的突变与视网膜色素变性有关。据我们所知,在任何PAR中自然发生的突变尚未被发现,特定疾病状态下PAR糖基化状态的变化也了解不多。
不同蛋白酶在不同位点水解PAR的能力产生了独特的系带配体和活性构象,这可能会影响配体诱导的磷酸化和泛素化。GPCRs的磷酸化对β-arrestin招募十分重要。β-Arrestins是能够促进GPCR从G蛋白信号解耦联和内化(信号终止的重要过程)的多功能接头蛋白质。此外,β-arrestins作为支架蛋白能够促进各种MAP激酶信号的级联。
之前的研究表明,配体激活的特定GPCR磷酸化修饰的程度和位点不仅影响β-arrestin-GPCR相互作用的稳定性,也指导β-arrestin特定功能的活性。这种现象被称为条形码假说。这个假说建立在如下两个事实之上:不同配体激活的β-肾上腺素受体磷酸化也不同,不同组织中相同配体激活的M3毒蕈碱受体磷酸化表达也不同。这都导致不同的β-arrestin功能。尽管PAR1和PAR2在活化后被大量磷酸化,但还没有研究检测到PAR在不同蛋白酶激活时的差异磷酸化及其对细胞行为的影响。
与磷酸化类似,许多GPCRs包括PAR1和PAR2在翻译后会被棕榈酰化和泛素化修饰。棕榈酰化是通过pal(一种16碳饱和脂肪酸)与半胱氨酸残基的共价连接来实现的。PAR1和PAR2与其他GPCR一样,在近膜C -尾部半胱氨酸上进行棕榈酰化,这似乎促进了棕榈酰基团插入脂质双分子层。PAR2的棕榈酰化调节受体表达、激动剂敏感性、脱敏和内化。相比之下,PAR1的棕榈酰化作用对于酪氨酸分类基序的接头蛋白的正确识别很重要,因而对细胞表面保留适量受体应答凝血酶至关重要。除了转运外,PAR1棕榈酰化还调节受体- g蛋白偶联,这是在PAR1半胱氨酸突变体中观察到的:小分子抑制剂JF5需要第八螺旋棕榈酰化的形成才能封闭PAR1激活Gαq信号。然而,JF5抑制剂的作用并不局限于PAR1,趋化因子CCR4和5-羟色胺5-HT2A
GPCRs(含半胱氨酸残基)和一个假定的第八螺旋也同样被抑制。这些数据表明,某些GPCRs的棕榈酰化作用似乎可以产生一个胞内界面,可作为治疗开发的目标。
泛素是一种由76个氨基酸组成的小蛋白,可以被含有泛素结合域的蛋白结合。泛素的主要功能是提供活化的GPCRs从核内体转移到溶酶体降解的分选信号。这显然是激动剂激活PAR2的情况。然而,新的研究表明,某些GPCRs的泛素化作用可以招募到促进信号应答的接头蛋白。这在最近的凝血酶激活的PAR1中得到了证实。在凝血酶的激活下,PAR1通过K63连接泛素迅速泛素化,后者通过泛素结合Npl4锌指结构域促进TAB2的结合。然后TAB1招募至活化的PAR1- TAB2,诱导p38 MAP激酶的自磷酸化和活化,促进内皮屏障的破坏。嘌呤P2Y1受体的泛素化作用类似于促进内皮细胞中p38 MAP激酶的活化,但其他PAR1活化蛋白酶是否引起类似反应尚不清楚。因此,对一个给定的PAR,应答不同的激活蛋白酶,重要的是翻译后的修饰类型,可以用来确定驱动一个特定的生理过程的信号应答,这对药物治疗研发有着重要的意义。
PAR的二聚化和药物研发
PAR二聚对药物开发具有重要意义,因为大多数药物都是针对PAR单体开发的。因此,研究人员必须考虑PAR单体的药理学扰动将如何影响二聚体或高阶低聚体的受体。在这里,我们讨论的研究表明,在正常细胞和疾病状态下,PAR相互作用,形成二聚体,并可能形成高阶低聚物。
大多数细胞类型表达一个以上的PAR,这些PAR的活性可以通过相互作用来调节。这在血管系统细胞中表达的内源性PAR的研究中得到了最好的例证。在人类血小板中,PAR1与PAR4共同表达,而在小鼠血小板中PAR3和PAR4共同表达。凝血酶可结合并水解小鼠PAR3;然而,被切割的PAR3似乎不产生自主信号,而是作为辅助因子促进PAR4的高效切割和激活。同样,PAR1似乎可以促进人血小板中PAR4的切割和激活。PAR1和PAR4也被证明在人血小板中形成复合物,这有助于凝血酶有效激活血小板。与PAR3相比,活化的PAR4对能有效地与G蛋白结合,并通过持续和延迟的信号传递促进血小板聚集。辅助水解激活其他PAR的受体应该与之接近,可能两者以异源二聚体的形式相互作用。
在内皮细胞中,PAR1和PAR3都有表达,缺失PAR3会减弱凝血酶促进内皮屏障破坏的能力。这是因为PAR3可以调节PAR1优先传递信号给Gα13。APC可以切割PAR1和PAR3,促进内皮细胞、神经元和小鼠足细胞的细胞保护信号。此外,APC诱导PAR1-PAR3异质二聚体的形成,但其机制尚不清楚。二聚体的形成有助于诱导不同的细胞类型中PAR1和par3特异性的信号反应。
PAR2在内皮细胞中低水平表达,在炎症过程中升高。PAR2升高的条件下,凝血酶水解PAR1 N末端,暴露出配体域可以反式结合并通过分子间作用激活PAR2,从而引发与只有受体激活剂作用不同的信号应答。使用PAR1封闭抗体和交叉脱敏实验研究内皮细胞发现,PAR2通过凝血酶激活PAR1而激活。在最近的研究中发现,在脓毒症进展过程中PAR1会反式激活PAR2。在这些病理条件下,由于内皮细胞PAR2表达的增加,PAR1信号从血管破坏转变为血管保护。与这些研究一致,在细胞因子处理的内皮细胞中,PAR2的表达增强了凝血酶激活的PAR1的信号能力,这与单独使用PAR1有差异。共免疫沉淀、荧光共振能量转移和生物荧光共振能量转移研究为PAR1和PAR2形成二聚体提供了大量证据。此外,凝血酶激活PAR1-PAR2二聚体走的是β-arrestins和Rac1信号通路,而不是受体原体介导的G蛋白质和RhoA信号通路。这些研究表明,除了在PAR1 N末端的不同位点水解的不同蛋白酶外,其他受体特性也会影响信号传递,包括细胞膜微区区隔、翻译后修饰和二聚作用。
结论
尽管研究人员在体内PAR功能以及在体内和体外描述受体偏信号特性方面取得了相当大的进展,但FDA只批准了一种靶向PAR1原位结合位点的药物。因此,开发针对PARs的新药理学制剂,包括正构和变构调节剂,存在着前所未有的机会。正构配体和变构配体都可以与特定的GPCR相互作用,并通过偶联不同的信号效应子来促进偏倚信号。因此,需要大力设计小分子,以促进PAR偏倚信号的方式,这可能导致更好的临床疗效并减少不良副作用。事实上,PAR2小分子抑制剂GB88和PAR1 parmodulin ML161抑制剂阻断了部分但不是全部的受体介导的信号通路,前景广阔。此外,基于结构的药物设计是开发新一代PAR抑制剂的一种有吸引力和可信赖的方法,并且可行,最近确定的结合了vorapaxar的PAR1的高分辨率晶体结构验证了这一点。因此,快速发展的GPCR晶体学方法可能会导致在不久的将来其他PAR结构的解出,这可以补充其他已建立的高通量筛选方法。总之,这些不同的策略将帮助研究人员开发新的PAR药物,可能更有选择性、更有效、副作用更少。