阅读者:刘绵宇
文章来源:Genomic selection in domestic animals: Principles, applications and perspectives
摘要
本文阐述了基因组选择的原理,解释了影响其效率的主要因素和提出的不同模型的假设。解释了其在奶牛中被快速采用的原因,并讨论了其应 用于其他物种的条件。发展张望包括:选择新的性状和新的育种目标;采用基于因果变异(SNP)信息的稳健方法;基因型×环境相互作用的预测。
前言
几个世纪以来,家畜种类的选育依赖于动物自身的表型。在20世纪,随着选择指数理论,以及最佳线性无偏预测(BLUP-一种依赖于混合线性模型的更复杂的方法)的提出,可以利用候选个体的亲属表型信息来预测其“育种值” (Breeding value)。对于具有中度或高遗传力且易于记录的表型性状,利用上述方法非常有效。而对于那些测量困难或遗传力较低的性状,取得有效地进展则需要昂贵的表型测定投资。
在过去的25年中,一些数量性状基因座(即基因组的区域,可以解释性状的遗传方差的一部分)已经通过遗传标记进行了定位,为标记辅助选择(MAS)铺平了道路。遗传标记是多态性序列,通常没有生物学效应,但易于基因分型,因此广泛用于遗传研究。MAS方法对于具有简单遗传决定机制的性状是有效的,但在许多更复杂的情况下则令人失望。两个导致MAS低效率的主要原因是:
1)这些少量QTL解释的遗传方差不但有限且总是被过高估计;
2)在群体水平上标记和QTL之间的关联度(或连锁不平衡)低。
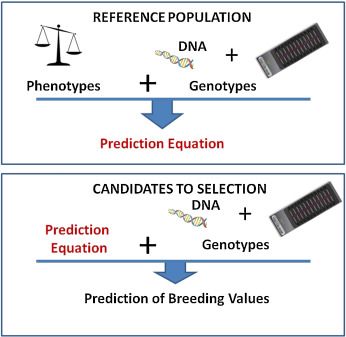
2001年,Meuwissen等人提出了一种新方法,利用覆盖全基因组的标记估计育种值。利用该方法,估计每个标记的遗传效应,然后累加用来预测每个动物的总体育种值(图1)。在参考群体(即同时具有表型和标记基因型信息的大量个体)内估计标记的效应,然后利用这些效应,对具有标记基因型信息但没有表型的候选个体进行选择。这种方法要想产生好的效果,要求基因分型的个体数量多,且覆盖基因组上的标记密度高。这就意味着在开发出大规模且廉价的基因分型方法之前,该方法难以实际应用。
1.奶牛基因组选择的成功
在基因组时代之前,奶牛的遗传改良依赖于分布在大多数法国农场的庞大的表型记录系统。通过长期的后裔测定,来筛选出最好的公牛。这些公牛通过人工授精的方式来繁殖下一代,根据产奶量和成分,对乳腺炎的抗性(即乳房感染),生育力,外形,产犊条件,寿命等大约40个性状,对每头公牛进行遗传评估。2007年,组装出第一个牛基因组草图后,llumina公司与国际联盟组织共同开发了一个芯片,可同时对54,000多个单核苷酸多态性(SNP)进行基因分型。虽然这些标记仅占所发现的SNP一小部分, 但是它们在很多品种中具有高度多态性并且在基因组上均匀分布。该芯片立即用于对现有且已经进行过后裔测试的公牛进行基因分析。利用这些最初的参考群体,基因组育种值已经足够精确可以取代后裔测试。在2009年,基因组选择被多个国家正式采用,允许分发只经过基因组评估的年轻公牛精液。
这是选择育种的革命性变化,后裔测试不再是必须的,简化了选择过程并降低了成本,由于大大减少了世代间隔,年遗传趋势可能翻倍,同时由于生产成本的降低,大量的奶牛将被选择和销售,进而可以更好地管理遗传资源,从遗传学上限制近交繁殖趋势,更容易满足多样化的需求和目标,选择更平衡和可持续的目标变得更容易,其中就包括低遗传力性状,如生育力或乳房炎抗性。
为提高育种值的准确性,需要构建大的参考群体,因此在育种合作社之间开始了国际合作,产生了大型合作组织,如针对荷斯坦奶牛的欧洲基因组学研究(九个欧洲国家)。为了降低基因分型成本,设计了一种低密度芯片,具有非常好的填充准确性,可以很好地预测缺失的标记。这产生了一个良性循环:大量已分型的个体降低了基因分型的成本,从而进一步增加基因分型个体的数量。
在农场一级,此工具现在用于优化畜群选择、交配和替换,因为每个基因分型的母牛和人工受精的公牛一样,都已经被准确评估。2015年12月,法国国家数据库包括400,000万头基因分型的牛,仅2015年就有10万头。现在,大量分型的奶牛(母牛)是群体替换和更新的主要参考资源。
在一些小的奶牛品种或很少人工授精公牛的肉牛品种中,参考群体部分由具有表型数据的母牛组成,是实施基因组选择的唯一方法。2016年,12个法国牛品种,包括几个小品种,在其育种计划中使用基因组选择。这是一个至关重要的变革,因为最初只有最大的品种才能从这项创新中受益,而小品种很难受益。
3. 影响基因组选择效率的因素
无论是那种家畜,每年的遗传增益取决于四个参数:性状的遗传变异,选择强度,评估准确性和世代间隔。基因组选择可以修改后三者。基因组选择的主要优点是可以对没有自身表型信息的候选个体进行评估和选择,不再需要其后代的信息。
因此,选择可以提早开展,刚出生后甚至在胚胎期就可以。在许多物种和生产系统中,是否开展基因组选择,取决于其能够在多大程度上减少世代间隔。当分型成本很低时,可以筛选大量的候选个体,可以增加选择强度。这种大规模筛选还可以更好地利用现有的遗传资源。可以对参考群体中记录的任何性状进行基因组选择评估,对于那些在候选个体上难以记录或者说不可能记录的性状(性别连锁性状,肉质性状,抗病性等),基因组选择具有优势。
选择效率的第三个决定因素,基因组评估的准确性(r),对于一个给定性状,取决于(1)对SNP效应估计的准确性和(2)SNP与因果变异之间的连锁不平衡。第一个参数取决于参考群体的大小(N )和性状的遗传力()。第二个参数取决于基因组的结构和性状的遗传结构。一个关键参数是在群体中分离的独立区段的数量。该数字q是基因组长度(长度= L,摩尔根量级)和有效群体大小Ne的函数。当Ne较小时,保守区段较长,并且需要较少的标记来跟踪它们。在实践中,通常使用公式来评估该准确度:,其中q是Ne和L的函数。
4.评估方法
已经建立了很多方法来进行基因组评估。由于标记数量众多,所有方法都将标记的效应视为一个随机效应,其值来自于一个先验统计分布,该分布根据方法而不同。从概念上讲,统计模型要么明确地包括标记的效应,要么利用基于标记信息的协方差结构,直接描述所有分型个体的基因组育种值。这两种模型完全等效,但根据不同的情形,选择其中一种可以更方便地评估或解释。基因组最佳线性无偏预测(或GBLUP),是多基因BLUP的简单扩展,其中亲缘关系矩阵基于标记信息代替系谱。
对于GBLUP方法,所有标记具有相同的权重;该模型忽略了性状的真实遗传决定机制,两个个体基因组育种值之间的协方差是与它们共有的基因组百分比成比例的。GBLUP对于真正由多基因决定的性状特别有效。其他评估方法旨在选择最具预测性的标记,推测其位于因果变异附近。已经提出了许多贝叶斯方法,这些方法赋予那些位于因果变异附近的SNP标记更大的权重或假设只有一小部分变异具有非零效应值。换句话说,它们试着更好地考虑QTL信息(这些方法对于多标记QTL定位也非常有效)。
在大多数方法中,评估方法单独处理每个个体的每个SNP,忽略它们的连锁不平衡。单倍型(由相邻SNP的组合定义)比双等位基因SNP更具信息量,并且更好地反映了血缘同源的情况。在法国奶牛中使用了一种方法,模型包括由性状关联的SNP单倍型追踪到的数千QTL,以及数千个SNP,它们以与GBLUP相当的方式量化剩余的残留多基因项。
这种综合模型可与其他方法一样准确且更加稳健,但代价是更高的复杂性。利用因果变异,将来该模型还可以进一步演化:当完全已知时,因果变异可以容易地替换作为其代理的单倍型。
在奶牛的第一次开创性工作之后,基因组选择正在越来越多的动物和植物种群和物种中使用。尽管如此,它仍然是一项非常新的创新,预计在不久的将来会有许多变革。
5 .基因组选择2.0
在奶牛的第一次开创性工作之后,基因组选择正在越来越多的动物和植物种群和物种中使用。尽管如此,它仍然是一项非常新的创新,预计在不久的将来会有许多变革。
5.1 扩展到许多种群
在大多数物种中,基因组选择的成本仍限制它的普及。许多育种计划无法承担建立一个参考群体的投资,并且基因分型成本仍然是具体实施的一个重要障碍;一个关键参数是基因分型与表型测定成本的比率。在高繁殖力的物种中,用非常低密度的芯片进行基因分型在经济上更为划算:当选择核心群中的所有亲本都以高密度进行基因分型时,只需要几百个标记就可以追踪候选个体(它们的后代)中的所有染色体片段,降低整体基因分型成本。这个例子表明,奶牛的情况不能简单地复制。基因组选择必须要根据生物学和经济条件进行设计
5.2 使用生物信息和因果变异的稳健方法
已经观察到基因组选择效率强烈依赖于待选群体和参考群体之间的亲缘关系。对于给定的性状,如果已知因果多态的遗传关系,则选择效率将是最大的。实际上,当前仅使用基于遗传标记的代理。当亲缘关系较近时,此代理更准确,而对于几乎没有亲缘相关的个体,此代理变得非常不准确。
此外,参考群体中估计的标记效应反映了该群体中的标记-QTL关联。当参考群体与候选群体之间的世代数增加时,QTL和它们周围的标记之间的重组事件累积,导致效率降低。因此,如果使用因果变异或非常近的标记,预计基因组评估对于缺乏近亲缘关系情况会更加稳健。
出于相同的原因,目前的基因组预测方法对于跨品种选择也不是有效的。在一个品种中建立的预测方程在另一个品种中几乎没有预测能力,除非品种间具有密切的亲缘关系。这在最初被解释为标记密度太低,导致参考群体中观察到的QTL-标记连锁不平衡,在候选群体无法保存。但是使用更高的密度(每4 kb或0.004 cM有一个标记的高密度芯片)并没有改善这种情况。
对通过对牛的模拟表明,只有使用非常接近的标记(在相同的千碱基间隔内),才能获得近似于因果变异水平上的跨品种基因组亲缘系数,而基因组上其他较远的标记会产生干扰,必须排除在外。当然,效率将取决于不同群体中QTL分离的比例及其效应的稳定性。这些观点仍在探讨中。然而,对于小品种来说,跨品种选择可能最有吸引力,因为可以组装足够大的参考群体。
经过最初的黑箱策略后,相信现在通过整合生物学知识,基因组评估可以更准确和更持久。多个团队通过关联分析在全序列水平上发起了大规模的QTL定位项目,以鉴定大量的候选变异,无论是因果变异还是与因果变异非常接近的区域,甚至是解释低至1%的遗传方差的QTL。为基因组选择构建的参考群体,包括数万个体,可用于作图资源群体。
通过整合不同品种的结果可以进一步提高作图分辨率,因为不同品种间的连锁不平衡衰变必须比品种内更快。这些大型资源群体的完整序列不能直接获得,但可以准确的填充。这是“1000公牛基因组”国际计划的主要动机。2015年7月已有1682个全基因组序列可供使用。在这个项目中,测序公牛主要是从其品种中选择对群体影响最大的祖先,以最大限度地增加填充准确性。
假设可以鉴定出许多因果变异,它们能够以一种简单的方式直接包括在基因组选择中。用于基因组选择的芯片中存在一个由用户设计的定制部分,包括这些候选变异。当这种芯片在商业群体中大规模使用后,可以确认这些候选变异的效应,并将它们整合到基因组预测模型中。
5.3 新的表型和新的育种目标
另一个预期的主要变革是选择性状和育种目标的灵活性更大。通过基因组选择,一旦建立了足够大小的参考群体后,性状改良的速度会非常快。在牛中,基于创新的记录技术(例如用于牛奶成分和牛奶特性的中红外光谱法)的使用,精确农业数据(健康,繁殖,行为...)的使用,商业数据(卫生卡,来自屠宰场的胴体特征)的使用或国际合作测量代价昂贵的性状(例如,饲料效率或甲烷排放),从而出现了不同的机会。目前正在采取许多举措来产生参考群,以改良那些长期以来被认为无法选择的性状。
6 结论
基因组选择在牛中非常成功,因为它以相似或更低的成本提供更多的遗传增益。但另一个重要的、常常被忽视的成果是,它为难以选择的性状、尚未选择但对可持续生产很重要的性状以及为长期更好地管理遗传变异提供了巨大的机会。
基因组选择是近期一项非常先进的创新。强有力的变革已经开始,包括降低基因分型成本,新性状的表型测定方法,参考群体的创建或替换方法,使用基因组序列鉴定的因果突变增加基因组预测的稳健性和持久性,或遗传×环境相互作用的基因组预测等。