目前单细胞测序获取细胞的方法主要有两种:
(1)Smart-seq:测序细胞量少(因为一次通过一个细胞),但是测序的reads数多;
(2)Drop-seq(10X ):可以测许多细胞,但是测得深度不深。
前些天练习了smartseq2的单细胞测序流程。这次就来练习一下10×测序的分析。
这次要练习的文章:
Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA(2018年9月发表在NC)。文章的具体解析在这里:爱恨难分—癌症免疫治疗获得性抗性。
这篇文章的测序样品来自两名患者(单细胞实战(一)数据下载):
患者2586-4:利用10X 3' Chromium v2.0平台建库 + Hiseq2500 "rapid run"模式。
(1)discovery tumor部分:After sequence alignment and filtering, 7431 tumor cells (2243 cells before and 5188 cells after T cell therapy)
(2)discovery PBMC部分:After sequence alignment and filtering, a total of 12,874 cells were analyzed [其中包含了四个时间点:治疗前(Pre),治疗后早期day +27(Early),治疗后反应期day+37(Resp),治疗后复发+614 (AR)]
患者9245-3:利用10X 5' V(D)J 进行cell washing, barcoding and library prep+ NovaSeq 6000(gene expression) + Hiseq4000 (V(D)J)。
The second validation patient (9245-3) is a 59-year-old man with metastatic MCC that had initially presented as stage IIIB disease, now metastatic at multiple sites.
(一)数据下载,提取fastq文件
首先下载原始数据。刚开始我在EBI上直接下载的fastq文件,可是看到教程里说后续的cell ranger软件分析需要输入两个文件。所以我又改下了sra文件(下载的方法就不说了,我的前一篇单细胞测序实战的文章里有提到如何批量下载),用fastq-dump提取fastq文件,这样就可以在使用fastq-dump时加参数,把read和UMI+Barcode文件分开来了。
#!/bin/bash
for i in SRR77229*
do
echo $i
fastq-dump --gzip --split-files /media/yanfang/FYWD/RNA_seq/sra_files/patient2586/$i
done

然后简单的看一下_1,_2,_3文件里都是写什么:
$ zless -SN SRR7722937_1.fastq.gz | head
@SRR7722937.1 SN367:911:HKMNCBCXY:1:1103:1119:1866 length=8
TTTCATGA
+SRR7722937.1 SN367:911:HKMNCBCXY:1:1103:1119:1866 length=8
GGGGGIII
@SRR7722937.2 SN367:911:HKMNCBCXY:1:1103:1182:1935 length=8
TTTCATGA
+SRR7722937.2 SN367:911:HKMNCBCXY:1:1103:1182:1935 length=8
AAAAGGGG
@SRR7722937.3 SN367:911:HKMNCBCXY:1:1103:1398:1864 length=8
TTTCATGA
$ zless -SN SRR7722937_2.fastq.gz | head
@SRR7722937.1 SN367:911:HKMNCBCXY:1:1103:1119:1866 length=26
AGCAGCCGTGACTACTGTATTGCGAC
+SRR7722937.1 SN367:911:HKMNCBCXY:1:1103:1119:1866 length=26
AGGGGIIIIIGGIIIIIIIIIIIIGG
@SRR7722937.2 SN367:911:HKMNCBCXY:1:1103:1182:1935 length=26
GCTCCTAAGACACTAAGGCCTGTACC
+SRR7722937.2 SN367:911:HKMNCBCXY:1:1103:1182:1935 length=26
A_1文件里每条序列8bp,_2文件里每条序列26bp,_3文件里每条序列98bp,这篇文献用的测序平台是10X Genomics 3' Chromium v2.0。那就要先知道这个平台的文库组成是什么样的。根据教程单细胞实战(二) cell ranger使用前注意事项:

这里面的read2长度不固定,有时会短几bp。Barcode用来标记细胞和细胞计数。UMI就是Unique Molecular Identifier,由4-10个随机核苷酸组成(所以上面的fastq文件里_1就是UMI),在mRNA反转录后,进入到文库中,每一个mRNA随机连上一个UMI(标记转录本),结果可以计数不同的UMI,最终统计mRNA的数量,这样与参考基因组比对后就可以定量基因的表达量 (转录本数量,近乎绝对定量)。那测序时需不需要加入标准物(如ERCC)的时候,官方给出的建议是不建议加ERCC(考虑到成本和影响细胞和基因的计数)。
根据教程里说的,我们需要把几个文件名改一下,方便后续的分析。
# 将原来的_1.fastq.gz改成_S1_L001_I1_001.fastq.gz
# 依次类推,将原来_2的改成R1,将_3改成R2。批量处理,就利用下载SRA的SRR ID好了。
cat SRR_Acc_List.txt | while read i ;do (mv ${i}_1*.gz ${i}_S1_L001_I1_001.fastq.gz;mv ${i}_2*.gz ${i}_S1_L001_R1_001.fastq.gz;mv ${i}_3*.gz ${i}_S1_L001_R2_001.fastq.gz);done
(ID如何下,请参考教程单细胞实战(一)数据下载 )
(二)了解Cell Ranger的使用
首先,Cell Ranger是什么? 官网的回答:Cell Ranger is a set of analysis pipelines that process Chromium single cell 3’ RNA-seq output to align reads, generate gene-cell matrices and perform clustering and gene expression analysis.也可以看这篇文章的介绍(10X单细胞测序分析软件:Cell ranger,从拆库到定量):
cellranger是10X genomics公司为单细胞RNA测序分析量身打造的数据分析软件,可以直接输入Illumina 原始数据(raw base call ,BCL或FASTQ)直接进行文库拆分、细胞拆分、输出表达定量矩阵、降维(PCA),聚类(Graph-based& K-Means)以及可视化(t-SNE)结果,结合配套的Loupe Cell Browser给予研究者更多探索单细胞数据的机会。
其次,这个软件能干点啥? 教程的答案(单细胞实战(三) Cell Ranger使用初探):
它主要包括四个主要基因表达分析流程:
(1)cellranger mkfastq : 它借鉴了Illumina的bcl2fastq ,可以将一个或多个lane中的混样测序样本按照index标签生成样本对应的fastq文件
(2)cellranger count :利用mkfastq生成的fq文件,进行比对(基于STAR)、过滤、UMI计数。利用细胞的barcode生成gene-barcode矩阵,然后进行样本分群、基因表达分析
(3)cellranger aggr :接受cellranger count的输出数据,将同一组的不同测序样本的表达矩阵整合在一起,比如tumor组原来有4个样本,PBMC组有两个样本,现在可以使用aggr生成最后的tumor和PBMC两个矩阵,并且进行标准化去掉测序深度的影响
(4)cellranger reanalyze :接受cellranger count或cellranger aggr生成的gene-barcode矩阵,使用不同的参数进行降维、聚类
它的结果主要是包含有细胞信息的BAM, MEX, CSV, HDF5 and HTML文件。
(三)下载安装cellranger,并下载10×Genomics所用的参考基因组信息
课程里写的是为了重复文章,所以他下载了和作者一样的cellranger2.0版本和2.1版本。然而,我根据课程的链接,并不能下载到这两个版本。。。所以我索性就下载了最新版3.1。主要目的是走一个流程,版本对我来讲无所谓。
(1)下载cellranger3.1版
去官网下载:https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
$ curl -o cellranger-3.1.0.tar.gz "http://cf.10xgenomics.com/releases/cell-exp/cellranger-3.1.0.tar.gz?Expires=1572780358&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cDovL2NmLjEweGdlbm9taWNzLmNvbS9yZWxlYXNlcy9jZWxsLWV4cC9jZWxscmFuZ2VyLTMuMS4wLnRhci5neiIsIkNvbmRpdGlvbiI6eyJEYXRlTGVzc1RoYW4iOnsiQVdTOkVwb2NoVGltZSI6MTU3Mjc4MDM1OH19fV19&Signature=DtuIA6Dyzv22DB7LfJAqlWhytmcAEHgEVzSR9YgQnkbW91MkFvorTSoOW2wFjRtF9NytOUdeA4M~oFci9su1Yh-3ZTKR27~qaZ-neywY96OppGtqcsgyr6iiWi91bICHOvkD5pVUrVsf0-u5rz9kd0NxltWCoEeVz8EECsocEnlYDL5SG1F1ykFn~WxQPvgX67puz816A0cN69jRDHMdzR4d3mVxj72SmXT193Y5r0UF0Kamyqovb1JRLAl~bz6Km8fomjBOSXlwGQIFllzAyjRHbQPCmisE3YnoetCW1sRl1-yDtYoRp1N16fP~qP8t8pWP-XEtBlhMFNR-~ZGSeg__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
(2)基因组下载
对于cellranger所用的基因组,官方是这样给的说明:Cell Ranger references are typically backwards compatible. It is not necessary to use a reference version that matches your Cell Ranger version.也就是说,这个3.1版本的cellranger可以兼容之前所有版本使用的基因组版本,不需要非要和3.1版本对应。所以我下载了一下3.0版本的基因组信息。(强迫症的童鞋可以去下载同样版本的基因组哈!)
人类GRCh38:
$ wget http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-GRCh38-3.0.0.tar.gz
或者
$ curl -O http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-GRCh38-3.0.0.tar.gz #这个貌似快一些
#解压
$ tar -xzvf refdata-cellranger-GRCh38-3.0.0.tar.gz
人类hg19:
$ wget http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-hg19-3.0.0.tar.gz
或者
$ curl -O http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-hg19-3.0.0.tar.gz
#解压
$ tar -xzvf refdata-cellranger-hg19-3.0.0.tar.gz
小鼠:
$ wget http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-mm10-3.0.0.tar.gz
或者
$ curl -O http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-mm10-3.0.0.tar.gz
#解压
$ tar -xzvf refdata-cellranger-mm10-3.0.0.tar.gz
ERCC下载:
$ curl -O http://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-ercc92-1.2.0.tar.gz
安装cellranger:
#先解压
$ tar zxvf cellranger-3.1.0.tar.gz
#添加环境变量
$ echo 'export PATH=/media/yanfang/FYWD/RNA_seq/cellranger/cellranger-3.1.0:$PATH' >> ~/.bashrc
#试一下看看是不是安装成功了
$ cellranger
/media/yanfang/FYWD/RNA_seq/cellranger/cellranger-3.1.0/cellranger-cs/3.1.0/bin
cellranger (3.1.0)
Copyright (c) 2019 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Usage:
cellranger mkfastq
cellranger count
cellranger aggr
cellranger reanalyze
cellranger mat2csv
cellranger mkgtf
cellranger mkref
cellranger vdj
cellranger mkvdjref
cellranger testrun
cellranger upload
cellranger sitecheck
上面我们下载的基因组信息,解压以后可以看一下几个子文件里都有什么:
#举个栗子,GRCh38的文件夹
$ tree
.
├── fasta
│ ├── genome.fa
│ └── genome.fa.fai
├── genes
│ └── genes.gtf
├── pickle
│ └── genes.pickle
├── reference.json
└── star
├── chrLength.txt
├── chrNameLength.txt
├── chrName.txt
├── chrStart.txt
├── exonGeTrInfo.tab
├── exonInfo.tab
├── geneInfo.tab
├── Genome
├── genomeParameters.txt
├── SA
├── SAindex
├── sjdbInfo.txt
├── sjdbList.fromGTF.out.tab
├── sjdbList.out.tab
└── transcriptInfo.tab
4 directories, 20 files
这里要注意的是,这里下载的GTF文件和我们通常下载的GTF是不一样的,这里的GTF文件是只包含有注释的基因类型。
(四)cellranger的使用
Cell Ranger主要的流程有:拆分数据 mkfastq、细胞定量 count、定量组合 aggr、调参reanalyze(单细胞实战(四) Cell Ranger流程概览 )。
(1)mkfastq 拆分数据
由于这篇文献作者没有上传最原始的BCL文件,所以我们没有办法练习BCL文件转化成fastq文件。一般是利用mkfastq或者bcl2fastq将BCL转成fastq文件。在教程(CellRanger走起(四) Cell Ranger流程概览)里有一些介绍,可以看看。
(2)count 细胞定量
这个过程要做的是细胞和基因的定量,过程包括比对、质控、定量。
先来看看使用方法:
$ cellranger count
/media/yanfang/FYWD/RNA_seq/cellranger/cellranger-3.1.0/cellranger-cs/3.1.0/bin
cellranger count (3.1.0)
Copyright (c) 2019 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Usage:
count
--id=ID #这个ID是指输出文件存放目录名
[--fastqs=PATH] #fastq文件储存的位置
[--sample=PREFIX] #sample要和fastq文件的前缀中的sample保持一致,作为软件识别的标志
--transcriptome=DIR #参考基因组存放位置
[options]
count [options]
count -h | --help | --version
利用了STAR比对的方法,可以看一下教程对于STAR比对的简单介绍(CellRanger走起(四) Cell Ranger流程概览):
这款比对工具比对速度快,灵敏度高,是ENCODE、GATK推荐使用的工具,允许基因的可变剪切。比对完之后,利用GTF文件将reads溯源回外显子区、内含子区、基因间区:如果一条read的50%以上与外显子有交集,那么就认为它在外显区;如果不在外显子区,与内含子有交集,那么就认为它在内含子区;与外显子、内含子都没有交集,那么就认为在基因间区。
然后就开始按照格式把自己的文件储存位置加上,比如patient2586的第一个样品SRR7722937:
$ cellranger count --id=patient2586 \
--fastqs=/media/yanfang/FYWD/RNA_seq/fastq_files/patient2586 \
--transcriptome=/media/yanfang/FYWD/RNA_seq/ref_genome/10_genomic_scRNA_refgenome/GRCh38/refdata-cellranger-GRCh38-3.0.0 \
--sample=SRR7722937 \
--nosecondary \
--jobmode=local
# nosecondary 只获得表达矩阵,不进行后续的降维、聚类和可视化分析(因为后期会自行用R包去做)
#jobmode:using 90% of available memory and all available cores
代码写好,可以运行了。但是cell ranger软件运行对电脑的配置是有要求的。。。

这是官网的说明。然而我的电脑内存只有8G,而且我没有服务器,距离最低要求64G差的太多了。我尝试着运行了一下:
2019-11-03 22:30:23 [runtime] (ready) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.DISABLE_FEATURE_STAGES
2019-11-03 22:30:23 [runtime] (run:local) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.DISABLE_FEATURE_STAGES.fork0.chnk0.main
2019-11-03 22:30:23 [runtime] (ready) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.CHEMISTRY_DETECTOR.DETECT_CHEMISTRY
2019-11-03 22:30:23 [runtime] (run:local) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.CHEMISTRY_DETECTOR.DETECT_CHEMISTRY.fork0.chnk0.main
2019-11-03 22:30:23 [runtime] (ready) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.SC_RNA_ANALYZER.CHOOSE_DIMENSION_REDUCTION
2019-11-03 22:30:23 [runtime] (run:local) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.SC_RNA_ANALYZER.CHOOSE_DIMENSION_REDUCTION.fork0.chnk0.main
2019-11-03 22:30:24 [runtime] (chunks_complete) ID.patient2586.SC_RNA_COUNTER_CS.SC_RNA_COUNTER.DISABLE_FEATURE_STAGES
然而只能跑一小会儿,然后就一直不动了,我只尝试了一个样,过夜跑还是停留在这个状态。。。我问了王院长,他说现在的10×单细胞测序一般公司可以把最后的count矩阵给你,不需要自己跑了。(我也不知道是真的假的,也有可能是他老板比较有钱?)总之要确保自己的电脑达到了cell ranger的最低配置要求。
虽然不能跑,但是还是来简单的了解一下输出的文件都有哪些(10X单细胞测序分析软件:Cell ranger,从拆库到定量):
输出文件:
.outs
├── analysis【数据分析文件夹】
│ ├── clustering【聚类,图聚类和k-means聚类】
│ ├── diffexp【差异分析】
│ ├── pca【主成分分析线性降维】
│ └── tsne【非线性降维信息】
├── cloupe.cloupe【Loupe Cell Browser 输入文件】
├── filtered_feature_bc_matrix【很重要,Seurat, Monocle的输入文件】
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── filtered_feature_bc_matrix.h5【过滤掉的barcode信息HDF5 format】
├── metrics_summary.csv【CSV format数据摘要】
├── molecule_info.h5【aggregate的时候会用到的文件】
├── raw_feature_bc_matrix【原始barcode信息】
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── possorted_genome_bam.bam【比对文件】
├── possorted_genome_bam.bam.bai【索引文件】
├── raw_feature_bc_matrix.h5【原始barcode信息HDF5 format】
├── web_summary.html【网页简版报告以及可视化】
└── *_gene_bar.csv_temp【过程文件】
(3) 构建Aggregation CSV
(4) 运行aggr
当实验中用到了多个GEM well,并且想放在一起分析时,需要先用cellranger count分析各个来自于一个GEM well的fastq文件 (与是否同一个lane测序没关系),然后再用cellranger aggr进行整合。
什么是 GEM Wells?
看这篇文章是怎么解释的:(10X单细胞测序分析软件:Cell ranger,从拆库到定量)每个GEM well是10X芯片上的一个单独的区室,从barcode池 (barcode whitelist,前面--qc的结果中有,评估barcode测序准确度,进而影响细胞鉴定准确度)中随机获取barcode用于标记细胞。为了保证整合多个文库时barcode不发生冲突,通常会在barcode后面加一个数字,标记其来源的GEM well,如AGACCATTGAGACTTA-1和AGACCATTGAGACTTA-2,barcode序列一样,但来源于不同的GEM well,也是不同的细胞。
这两步我没办法运行,但是这里的课程(CellRanger走起(四) Cell Ranger流程概览)里对于这两步有具体的代码,可以参考。也可以直接去官网(https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate)里也有非常详细的步骤和代码。对于有条件的童鞋,在运行完上面的步骤后,可以参考教程(10X scRNA免疫治疗学习笔记-2-配置Seurat的R语言环境)来进行后续的分析。下面我会直接使用作者上传的原始矩阵进行后续的练习。
(五)下载文章的原始矩阵,并利用Seurat进行后续分析
下载地址在这里:patient2586

先练习PBMC这个矩阵,练习的流程按照教程走(10X scRNA免疫治疗学习笔记-3-走Seurat标准流程)。
(1)载入数据
> rm(list = ls())
> options(warn=-1)
> suppressMessages(library(Seurat))
#在载入R包时候常常会出现一些消息,如版本或函数覆盖的消息。这些消息是不必要的,可以使用suppressMessages()来进行关闭。
#设置工作文件夹
> setwd("/media/yanfang/FYWD/RNA_seq/matrix/10×scRNA_analysis")
# 读取表达矩阵
> start_time <- Sys.time() #获取当前的系统时间,达到秒的精度
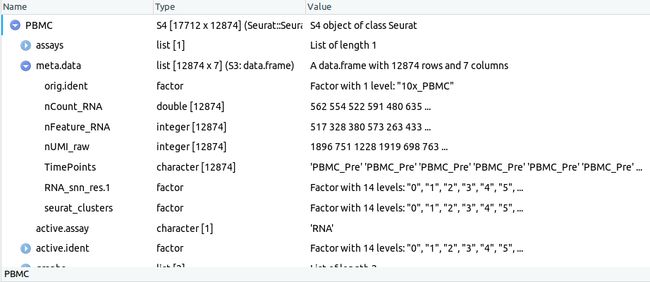
> raw_dataPBMC <- read.csv('./GSE117988_raw.expMatrix_PBMC.csv.gz', header = TRUE, row.names = 1)
> dim(raw_dataPBMC) #17712基因,12874细胞
(2)对文库大小进行归一化
> #sweep()里第一个要标注对哪个矩阵进行操作;第二个位置1和2跟apply一样的,2表示列;
> #第三个位置是要操作的向量,如果要对行操作,那么这个向量长度就要和行数一样
> #最后一个位置是计算符,比如:+ - * / < > 等
> dataPBMC <- log2(1 + sweep(raw_dataPBMC, 2, median(colSums(raw_dataPBMC))/colSums(raw_dataPBMC), '*'))
所以这里的操作是:先求每个细胞文库的总大小,然后用它的中位数除以总大小得到一个小数,然后按列乘以这个小数就相当于对文库进行了归一化,将文库本身的大小差异置之度外(10X scRNA免疫治疗学习笔记-3-走Seurat标准流程)。
NOTE:对于sweep(),还可以看看这篇文章:R中的sweep函数。
(3)划分时间点
在这篇文献里,作者使用了4个时间点。
> head(colnames(dataPBMC))
[1] "AAACCTGAGCGAAGGG.1" "AAACCTGAGGTCATCT.1" "AAACCTGAGTCCTCCT.1" "AAACCTGCACCAGCAC.1" "AAACCTGGTAACGTTC.1"
[6] "AAACCTGGTAAGGATT.1"
> tail(colnames(dataPBMC))
[1] "TTTGTCAAGCGAGAAA.4" "TTTGTCAAGGAATTAC.4" "TTTGTCAAGTGCGTGA.4" "TTTGTCACACGAGGTA.4" "TTTGTCATCATTGCGA.4"
[6] "TTTGTCATCCACGCAG.4"
这里的.1到.4分别对应着:
1 => PBMC_Pre
2 => PBMC_EarlyD27
3 => PBMC_RespD376
4 => PBMC_ARD614
然后把上面列名里.1.2.3.4分别提取出来,分成4个组:
> timePoints <- sapply(colnames(dataPBMC), function(x) unlist(strsplit(x, "\\."))[2]) #点号是正则匹配符,需要用\\来转义
> table(timePoints)
timePoints
1 2 3 4
2082 1592 4684 4516
把1,2,3,4对应到具体的时间点名称上:
#对应到具体的时间点名称上
> timePoints <-ifelse(timePoints == '1', 'PBMC_Pre',
ifelse(timePoints == '2', 'PBMC_EarlyD27',
ifelse(timePoints == '3', 'PBMC_RespD376', 'PBMC_ARD614')))
> table(timePoints)
timePoints
PBMC_ARD614 PBMC_EarlyD27 PBMC_Pre PBMC_RespD376
4516 1592 2082 4684
(4)对表达矩阵进行质控
第一步:过滤一个基因在多少细胞表达
# fivenum():返回五个数据:最小值、下四分位数、中位数、上四分位数、最大值。
#对于奇数个数字,fivenum()先排序,依次返回最小值、下四分位数、中位数、上四分位数、最大值
> fivenum(apply(dataPBMC,1,function(x) sum(x>0) ))
RP4-669L17.10 LAMB3 NAT10 AC093673.5 RPL21
1 6 50 207 12102
#结果显示有75%的基因在207个细胞里表达
第二步:每一个细胞里有多少个基因表达
> fivenum(apply(dataPBMC,2,function(x) sum(x>0) ))
CAAGAAATCGATCCCT.2 GGCCGATTCCAGAGGA.3 TCAACGAAGAGCTGGT.3 TGCCAAAGTCTGCGGT.4 TCTGGAATCTATCGCC.3
10 321 395 481 2865
#结果说明75%的细胞里只表达481个基因
一般来说:10X的数据应该能做到平均表达800个基因
#hist 用于绘制直方图,横坐标为不同的区间,纵坐标为落入该区间内的频数
> hist(apply(dataPBMC,2,function(x) sum(x>0) ))
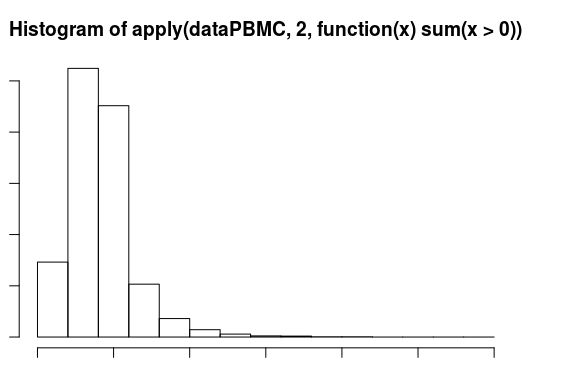
开始Seurat分析流程
(5)创建Seurat对象
> PBMC <- CreateSeuratObject(dataPBMC,
min.cells = 1, min.features = 0, project = '10x_PBMC')
> PBMC
An object of class Seurat
17712 features across 12874 samples within 1 assay
Active assay: RNA (17712 features)
(6)添加metadata
> PBMC <- AddMetaData(object = PBMC,
metadata = apply(raw_dataPBMC, 2, sum),
col.name = 'nUMI_raw')
> PBMC <- AddMetaData(object = PBMC, metadata = timePoints, col.name = 'TimePoints')
(7)聚类
标准流程:
标准化、找高变异基因、根据这些基因进行PCA降维、根据PCA结果找分群、TSNE降维、可视化。
NOTE:这里我安装的seurat是V3版本的,如果是V2版本的话,代码会有些许不同。在教程里有两个版本的代码,可以参考(10X scRNA免疫治疗学习笔记-3-走Seurat标准流程)。
#标准化
> PBMC <- ScaleData(object = PBMC, vars.to.regress = c('nUMI_raw'), model.use = 'linear', use.umi = FALSE)
#然后会弹出下面的进度条
Regressing out nUMI_raw
|=============================================================================================================| 100%
Centering and scaling data matrix
|=============================================================================================================| 100%
#找高变基因,这里Seurat V3里是FindVariableFeatures(), 而V2是FindVariableGenes()
> PBMC <- FindVariableFeatures(object = PBMC, mean.function = ExpMean, dispersion.function = LogVMR, mean.cutoff = c(0.0125,3), dispersion.cutoff = c(0.5,Inf))
Calculating gene variances
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Calculating feature variances of standardized and clipped values
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
# PCA降维
> PBMC <- RunPCA(object = PBMC, pc.genes = [email protected])
PC_ 1
Positive: FTL, FTH1, S100A9, GPX1, CST3, LYZ, S100A8, SAT1, AIF1, PPBP
LST1, FCN1, CTSS, CSTA, PF4, S100A12, GNG11, TYROBP, PSAP, NRGN
SDPR, VCAN, COTL1, LGALS2, S100A6, RP11-1143G9.4, MAP3K7CL, NEAT1, HIST1H2AC, RGS18
Negative: NKG7, IL32, GNLY, GZMA, HLA-A, UBB, CST7, MALAT1, GZMH, GZMB
CTSW, FGFBP2, KLRB1, CD7, BTG1, HOPX, CMC1, CD247, LTB, KLRD1
PRF1, CD3D, CXCR4, GZMM, TRBC2, TRBC1, TRAC, TRDC, CCL5, HBA1
PC_ 2
Positive: S100A9, LYZ, S100A8, AIF1, FCN1, CTSS, LST1, S100A6, TYROBP, FTL
CSTA, NEAT1, S100A11, S100A12, VCAN, LGALS2, RP11-1143G9.4, CST3, TYMP, PSAP
LGALS1, SERPINA1, DUSP1, S100A4, FOS, MNDA, VIM, MAFB, CD14, MS4A6A
Negative: PF4, GNG11, SDPR, HIST1H2AC, TUBB1, PPBP, ACRBP, RGS18, CLU, NRGN
GP9, SPARC, MAP3K7CL, NGFRAP1, NCOA4, TSC22D1, TREML1, MMD, PTCRA, RUFY1
PGRMC1, CLEC1B, C6orf25, HIST1H3H, CMTM5, ITGA2B, TMEM40, MYL9, TUBA4A, CCL5
PC_ 3
Positive: HBB, HBA1, HBA2, ALAS2, CD79A, AHSP, HBD, SNCA, IGKC, IGHD
SLC25A37, IGHM, HLA-DRA, TCL1A, CA1, CD79B, HBM, CD74, MS4A1, IGLC2
HLA-DQA1, SLC25A39, VPREB3, LTB, BLVRB, HLA-DRB1, BANK1, BPGM, LINC00926, FCER2
Negative: NKG7, B2M, CCL5, CST7, GNLY, GZMB, HLA-A, GZMA, FGFBP2, S100A4
MALAT1, HCST, GZMH, CTSW, ACTB, IFITM2, PRF1, KLRB1, KLRD1, ACTG1
CD247, GAPDH, CMC1, CD7, HOPX, KLRF1, SPON2, SRGN, TRDC, FCGR3A
PC_ 4
Positive: HBA1, HBA2, HBB, ALAS2, AHSP, HBD, SNCA, SLC25A37, CA1, HBM
BLVRB, UBB, SLC25A39, BPGM, MPP1, GMPR, HBQ1, S100A8, PDZK1IP1, S100A9
S100A12, NCOA4, LYZ, VCAN, IFI27, RP11-1143G9.4, TYROBP, MYL4, FOS, CSTA
Negative: CD74, CD79A, MALAT1, HLA-DRA, HLA-DPB1, CD37, IGHD, HLA-DRB1, BTG1, IGKC
CD79B, HLA-DPA1, HLA-DQA1, IGHM, TCL1A, CXCR4, B2M, CD52, MS4A1, IGLC2
LTB, HLA-DQB1, VPREB3, BANK1, PLPP5, FCER2, LINC00926, IGLC3, CD69, HLA-DOB
PC_ 5
Positive: TRAC, IL7R, CD3D, IL32, LTB, GZMK, CD52, TRBC2, VIM, CD8A
DUSP2, TRBC1, JUNB, MALAT1, B2M, S100A4, GAPDH, KLRG1, S100A12, TRGC2
HLA-A, S100A6, S100A8, VCAN, CXCR4, RP11-1143G9.4, BTG1, FOS, S100A9, MIAT
Negative: FCGR3A, HLA-DRA, GZMB, HLA-DPA1, HLA-DPB1, CD74, UBB, FGFBP2, HLA-DRB1, SPON2
TYROBP, FCER1G, PRF1, KLRF1, HLA-DQA1, NKG7, CD79A, CD79B, RHOC, HBA1
CST7, HBA2, ALAS2, HBB, HBD, IGHD, AHSP, IFITM3, CLIC3, IGFBP7
#根据PCA结果找分群,要分两步,而Seurat V2版本在这里只有一步
> PBMC <- FindNeighbors(PBMC, reduction = "pca", dims = 1:10,
k.param = 35)
Computing nearest neighbor graph
Computing SNN
> PBMC <- FindClusters(object = PBMC,
resolution = 1, verbose=F)
#需要注意的是,这里的resolution的赋值会影响后面的分群个数
#tSNE降维
> PBMC <- RunTSNE(object = PBMC, dims.use = 1:10)
这里提一句:
t-SNE中集群之间的距离并不表示相似度 ,同一个数据上运行t-SNE算法多次,很有可能得到多个不同“形态”的集群。
#可视化
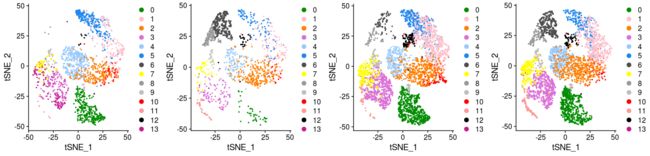
> DimPlot(PBMC, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black'))
Error: Insufficient values in manual scale. 14 needed but only 13 provided.
这一步我按照教程里的代码,输入了13个不同的颜色,结果报错,说需要14个颜色,少了一个。(之所以会少一个群是因为前面的resolution我用的是1,如果用0.9就会得到13个群)于是我就看了帮助文档,想查一下颜色的代号,帮助文档里说这个网站(http://colorbrewer2.org/#type=sequential&scheme=RdPu&n=3)可以挑选不同的颜色,并且有其代号。所以我挑了一个颜色以后,把代号加入到上面的代码里:
> DimPlot(PBMC, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a'))

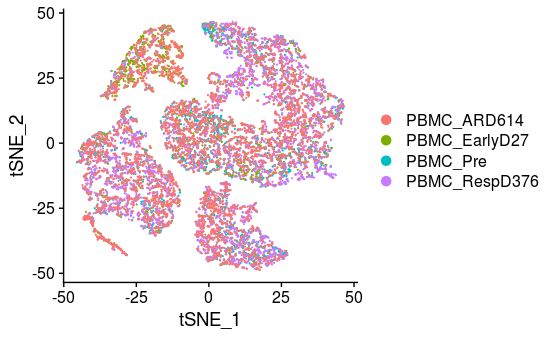
(8)看看作者整合的批次之间的差距
这一步就不像上面按照细胞群来做tSNE了,这回按照作者分的4个时间点来映射。理论上在所有时间点里应该有所有细胞类型。
> TSNEPlot(PBMC,group.by = "TimePoints")
> #用table对比一下
> table([email protected]$TimePoints,[email protected])
0 1 2 3 4 5 6 7 8 9 10 11 12 13
PBMC_ARD614 664 552 582 611 558 279 394 271 132 111 62 197 83 20
PBMC_EarlyD27 43 127 271 104 124 107 436 50 193 26 25 54 28 4
PBMC_Pre 370 184 218 105 476 332 1 65 3 47 77 8 10 186
PBMC_RespD376 806 747 527 717 339 459 90 285 71 202 220 68 135 18
(9)可视化marker基因
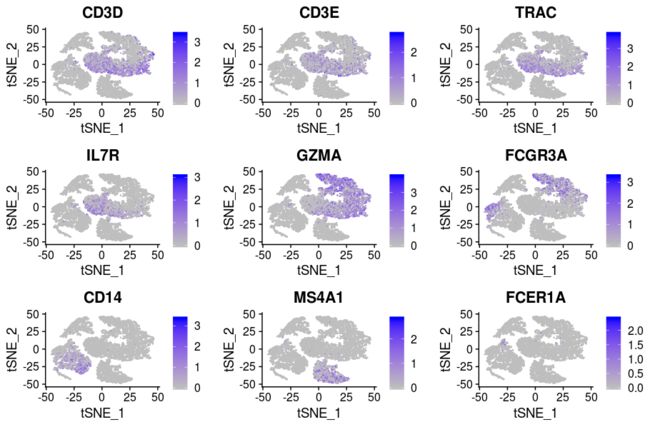
作者选取了一些基因,这些基因在不同的免疫细胞群里是特异表达的:
"CD3D","CD3E", "TRAC", "IL7R", "GZMA", "FCGR3A", "CD14", "MS4A1", "FCER1A" 。
$ markerGenes <- c(
"CD3D",
"CD3E",
"TRAC",
"IL7R",
"GZMA",
"FCGR3A",
"CD14",
"MS4A1",
"FCER1A"
)
> FeaturePlot(object = PBMC,
features =markerGenes,
cols = c("grey", "blue"),
reduction = "tsne")
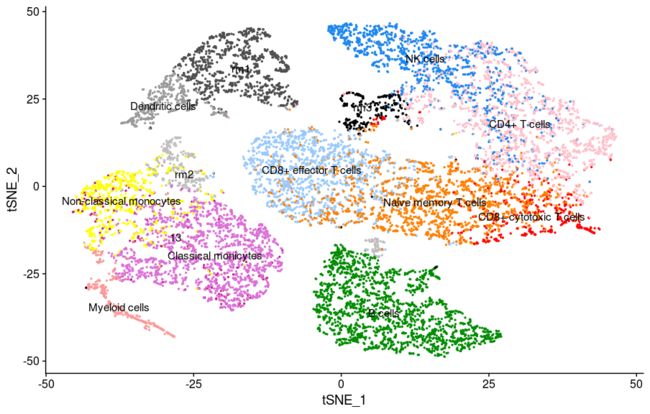
(10)把tSNE每一个分群加上细胞群名称
由于原文作者用的是Seurat v2进行分析,他得出了13个群,而我得到了14个分群,所以下面在命名的时候最后一个群我就直接以“13”命名(第一个群的编号是0)。
$ cat >celltype-patient1-PBMC.txt
0 "B cells"
1 "CD4+ T cells"
2 "Naive memory T cells"
3 "Classical monicytes"
4 "CD8+ effector T cells"
5 "NK cells"
6 "rm1"
7 "Non-classical monocytes"
8 "Dendritic cells"
9 "rm2"
10 "CD8+ cytotoxic T cells"
11 "Myeloid cells"
12 "rm3"
13 "13"
> names(new.cluster.ids) <- levels(PBMC)
> PBMC_V3 <- RenameIdents(PBMC, new.cluster.ids)
> DimPlot(PBMC_V3, reduction = "tsne", label = TRUE, pt.size = 0.5, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a')) + NoLegend()
(11)按不同时间点绘制分群结果
先得到每个时间点都有多少细胞:
> TimePoints = [email protected]$TimePoints
> table(TimePoints)
TimePoints
PBMC_ARD614 PBMC_EarlyD27 PBMC_Pre PBMC_RespD376
4516 1592 2082 4684
把不同时间点的细胞分别提取出来,这里要用到seurat包里的SubsetData这个函数。
先来看看这个SubsetData怎么用:
SubsetData(object, cells = NULL, subset.name = NULL,
low.threshold = -Inf, high.threshold = Inf, accept.value = NULL,
...)
object: 要先说明你要对哪个对象操作。
cells: 要说明你要提取这个对象里的哪些细胞。你可以指定列名,或者满足一定要求的细胞。如果这一项你不指定,将会按照下面三个参数的设置来提取满足要求的细胞。
subset.name: 要提取的子集的参数,例如:a column name in [email protected], etc.
low.threshold:参数的Low cutoff (default is -Inf)。
high.threshold:参数的High cutoff(default is Inf)。
。。。
#比如第一个子集,要取在PBMC里面meta.data里的timePoint这一项中PBMC_Pre这个时间点的细胞(下图有个截图,可以更直观的看到取的是什么
> PBMC_Pre= SubsetData(PBMC,cells=([email protected]$TimePoints =='PBMC_Pre'))
#然后将这个子集可视化,存入一个变量figure1
> figure1= DimPlot(PBMC_Pre, reduction = "tsne", label = FALSE, pt.size = 0.5, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a'))
#同理,把剩下的三个子集取出来,同样的处理
> PBMC_EarlyD27 = SubsetData(PBMC,cells=([email protected]$TimePoints =='PBMC_EarlyD27'))
> figure2=DimPlot(PBMC_EarlyD27, reduction = "tsne", label = FALSE, pt.size = 0.5, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a'))
> PBMC_RespD376=SubsetData(PBMC,cells=([email protected]$TimePoints =='PBMC_RespD376'))
> figure3=DimPlot(PBMC_RespD376, reduction = "tsne", label = FALSE, pt.size = 0.5, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a'))
> PBMC_ARD614=SubsetData(PBMC,cells=([email protected]$TimePoints =='PBMC_ARD614'))
> figure4=DimPlot(PBMC_ARD614, reduction = "tsne", label = FALSE, pt.size = 0.5, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black', '#c51b8a'))
然后我想把四个时间点的分群图并排的画在一起(因为文献里是同时展示了4张图,我之前也没试过,这次练习一下),要用到multiplot这个函数,这个函数是在Rmisc包(也有人说这个函数在ggplot2包里,但是我的ggplot2无法调用这个函数)。
> library(Rmisc)
> multiplot(figure1,figure2,figure3,figure4,cols=4)