关于SAM
Sam: I Want You!

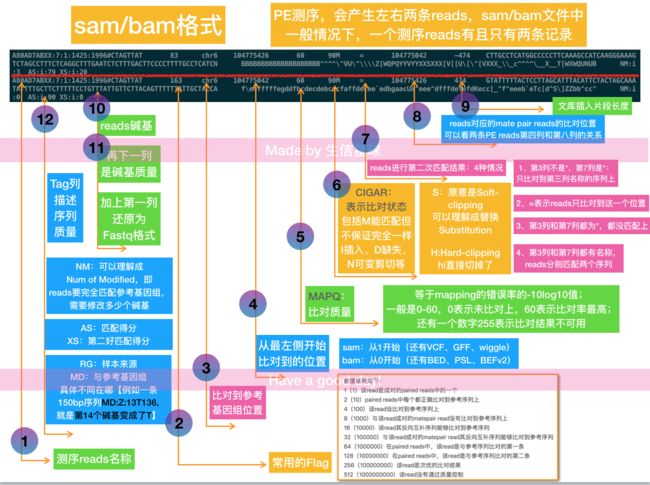
sam是短序列比对默认的标准格式,由sanger制定,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,另外也可以表示其他的多重比对结果。一般把测序reads比对到参考基因组以后,通常得到的就是sam文件,全称是sequence alignment/map format,BAM就是SAM的二进制文件(B即:Binary)
官方参考文档http://samtools.github.io/hts-specs/SAMv1.pdf
需要知道的名词
template:DNA/RNA序列的一部分,用它进行测序或者由原始数据拼接得到它,作为研究的模版;
【参考序列和比对上的序列共同组成的序列为template】
read:测序仪下机得到的原始数据,这些数据依照测序时间的先后被打上不同标签;双端测序存在read1,read2
segment:比对到read的时候的一段连续序列或者被分开几条子序列。一条read可以包含多条segment;
linear alignment:一条read比对到参考序列上,可以存在插入(insert)、缺失(delete)、跳跃(skip)、剪切(clip),但是方向不变(不能是一部分和正链匹配,另一部分又和负链匹配),sam文件中只占用一行记录;
chimeric alignment:“嵌合比对” 的形成是由于一条测序read比对到基因组上时分别比对到两个不同的区域,而这两个区域基本没有overlap。因此它在sam文件中需要占用多行记录显示。只有第一个记录被称作"representative",其他的都是"supplementary"【Chimeric reads are also called split reads】


read alignment:不管黑猫(linear alignment)、白猫(chimeric alignment),抓住老鼠(能够完整表示一个read)就是好猫(read alignment)
multiple mapping:假如有一个短序列,他在比对的时候看到哪哪都有他的影子,这种就是受重复区域影响比较大,所以read越长出现这种的可能性越低。一般指定首先匹配上的为最优匹配结果primary。
坐标系统:
- 1-based coordinate system :序列的第一个碱基设为数字1,如SAM,VCF,GFF,wiggle格式
- 0-based coordinate system :序列的第一个碱基设为数字0,如BAM, BCFv2, BED, PSL格式
SAM要处理好的问题:
- 非常多的序列(read),mapping到多个参考基因组(reference)上;
- 同一条序列,分多段(segment)比对到参考基因组上;
- 各种结构化信息表示,包括错配、删除、插入等比对信息;
具体的Sam格式:
花花之前有介绍,并且在Linux考试题中也有所提及
关于Samtools

1. View
-
sam转bam,-S指输入文件格式(不加-S默认输入是bam),-b指定输出文件(默认输出sam)
samtools view -Sb SRR3589956.sam >SRR3589956.bam -
【如果要bam转sam,-h设置输出sam时带上头注释信息】
samtools view -h SRR3589956.bam > SRR3589956.sam -
过滤功能-F:后接flag数字,常用有4(表示序列没比对上)、8(配对的另一条序列,即mate序列没比对上)以及12(两条序列都没比对上)。加上-F就表示过滤掉这些情况
如果是-f,就是提取指定flag的序列
2. Sort
samtools sort -@ 10 -o SRR3589956.sorted.bam SRR3589956.bam
3. merge
samtools merge xxx.merged.bam xxx.sorted.bam
4. index
samtools index SRR3589956.sorted.bam
插播一条
当有多个bam文件时,一般思路就是对每一个bam进行sort、index后,再merge成一个整体merged.bam,然后对merged.bam再进行sort、index,才算能用了,得到最终结果应该是是sorted.merge.bam
5. faidx
samtools faidx hg19.fasta
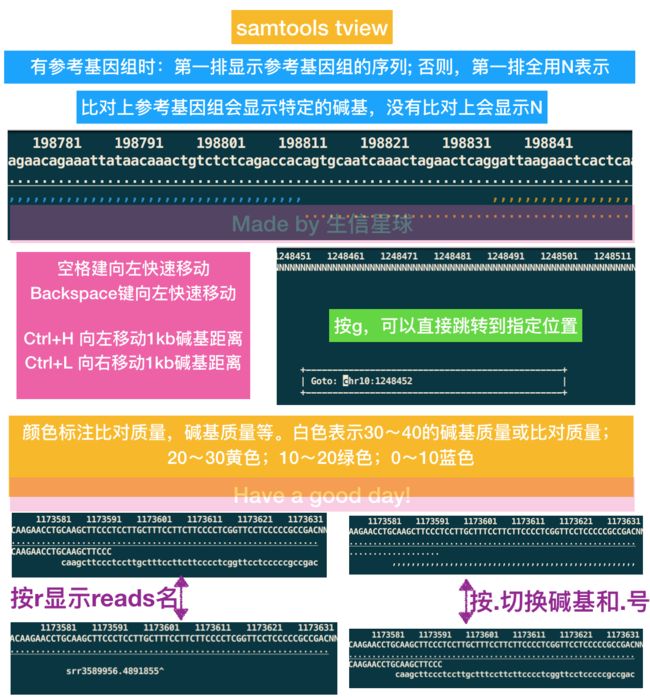
6. tview
samtools tview SRR3589956.sorted.bam hg19.fasta
7. flagstat
samtools flagstat SRR3589956.sorted.bam > SRR3589956.sorted.flagstat.txt
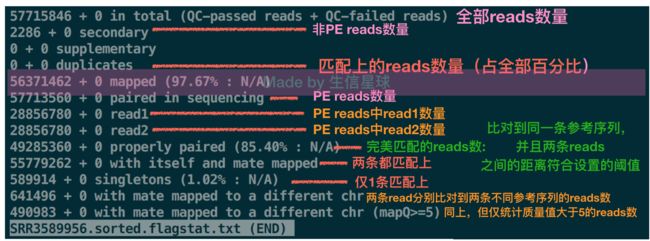
更新于19.7.5,最近收到细心小伙伴提醒,这个图中的secondary有点问题。可以参考:https://www.biostars.org/p/166444/,其中Devon Ryan 给出的答案是:A secondary alignment occurs when a given read could align reasonably well to more than one place. One of the possible reported alignments is termed "primary" and the others will be marked as "secondary".
8. depth
-r:(region)加染色体号;
-q:要求测序碱基质量最低值;
-Q:要求比对的质量最低值
samtools depth SRR3589956.sorted.bam >SRR3589956.depth
9. mpileup
-f:输入有索引的参考基因组fasta;
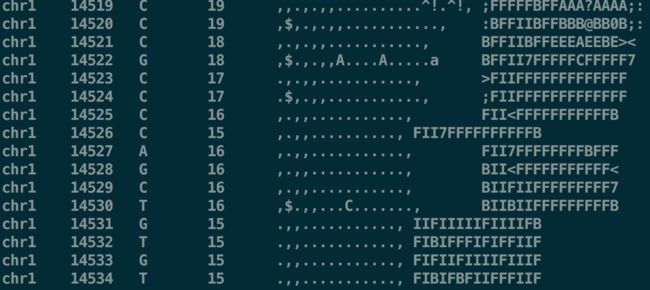
-g:输出到二进制的bcf格式【不使用-g,就不生成bcf格式,而是一个文本文件,统计了参考序列中每个碱基位点的比对情况;每一行代表参考序列中某一个碱基位点的比对结果】
samtools mpileup -f hg19.fa SRR3589956.sorted.bam > SRR3589956.mpileup.txt
结果文件大小 7.1G Aug 3 21:48 SRR3589956.mpileup.txt
结果包含6列:参考序列名、匹配位置、参考碱基、比对上的reads数、比对的情况、比对的碱基质量
10. bam转fastq
作用:方便提取出一段比对到参考序列的reads进行分析
利用软件:http://www.hudsonalpha.org/gsl/information/software/bam2fastq
11. rmdup
作用:将测序数据中由于PCR duplicate得到的reads去掉,只保留比对质量最高的reads
samtools rmdup默认只对PE数据进行处理
12. idxstats
作用:输出一个表格,包含“序列名、序列长度、比对上的reads数、没有比对上的reads数”;其中第四列指PE reads中的一条read能匹配到参考基因组的染色体A,另一条read不能匹配到A上
samtools idxstats
13. reheader
作用:替换bam文件的头文件
samtools reheader
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
