
valr: Reproducible genome interval analysis in R
- https://f1000research.com/articles/6-1025/v1
放在最前面的valr包的说明书:
- https://cran.r-project.org/web/packages/valr/valr.pdf
功能:
- 提取exon、intro、5'UTR、3’UTR等区间坐标
- 拓宽bed文件上下游或者body区间
- 生成启动子区间
- 取两个bed文件的交集、各自的补集、以及一个bed文件中去除两一个bed文件的坐标等
- 将bed文件分成若干个区间,
- 指定区间宽度
- 可以根据每个区间的长度,分别增加区间的百分比数量级
- 根据分成的若个bin,统计落在bin中的reads、DNA甲基化、未甲基化等的数目
- 随机从染色体区间生成若干个bin
- 可以对bed文件进行基因或者element的注释
- .....
总之这就是R中实现bedtools等工具的功能,有时间可以查看每个函数的源代码。如果您能掌握这里面每个函数的源代码,个人觉得在数据处理这一块几乎没有什么障碍了。
敲之前所需要理解的基因结构图
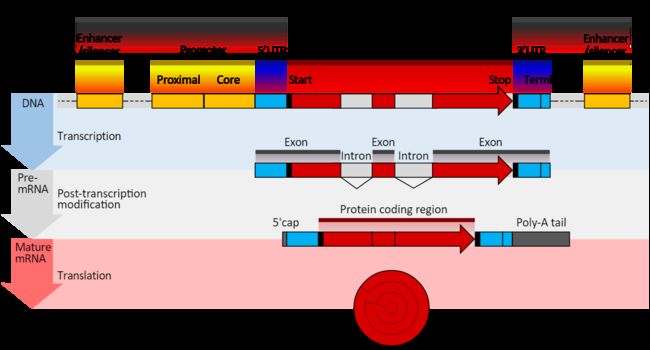
图片来源维基百科
- 模糊来说DNA包括Promoter、5'UTR、Open reading frame、3'UTR、Enhancer、silencer。
- exon(包括5'UTR 和 3'UTR)、intron
- CDS(conding sequnce)为去除5'UTR 和 3'UTR的exon区域,(所以以下CDS的起始位点并不是该基因的exon的起始和终止位点)
说明书代码: 逐行敲
# https://github.com/rnabioco/valr
# devtools::install_github('rnabioco/valr')
install.packages('valr')
library(valr)
library(dplyr)
#######################################################################
## as.tbl_genomeCoerce objects to tbl_genome.
#######################################################################
gr <- GenomicRanges::GRanges(seqnames = S4Vectors::Rle(c("chr1", "chr2", "chr1", "chr3"),
c(1, 1, 1, 1)),
ranges = IRanges::IRanges(start = c(1, 10, 50, 100),
end = c(100, 500, 1000, 2000),
names = head(letters, 4)),
strand = S4Vectors::Rle(c("-", "+"), c(2, 2)))
x <- as.tbl_interval(gr) ## 将其转换成tibble格式
# There are two ways to convert a tbl_interval to GRanges:
# 将数据框转换成GRanges格式
gr <- GenomicRanges::GRanges(seqnames = S4Vectors::Rle(x$chrom),
ranges = IRanges::IRanges(start = x$start + 1,end = x$end,names = x$name),
strand = S4Vectors::Rle(x$strand))
# or:
gr <- GenomicRanges::makeGRangesFromDataFrame(dplyr::mutate(x, start = start +1))
#######################################################################
## bed12_to_exonsConvert BED12 to individual exons in BED6.
#######################################################################
x <- read_bed12(valr_example('mm9.refGene.bed.gz'))
bed12_to_exons(x)
#######################################################################
## bed_absdistCompute absolute distances between intervals.
# 计算x中起始位置距离在y中最近的起始位置的距离
#######################################################################
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
x <- bed_random(genome, seed = 1010486)
y <- bed_random(genome, seed = 9203911)
bed_absdist(x, y, genome)
#######################################################################
## bed_closestIdentify closest intervals.
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,'chr1', 100, 125)
y <- trbl_interval(~chrom, ~start, ~end,'chr1', 25, 50,'chr1', 140, 175)
bed_glyph(bed_closest(x, y))
x <- trbl_interval(~chrom, ~start, ~end,"chr1", 500, 600,"chr2", 5000, 6000)
y <- trbl_interval(~chrom, ~start, ~end,"chr1", 100, 200,"chr1", 150, 200,"chr1", 550, 580,"chr2", 7000, 8500)
bed_closest(x, y)
bed_closest(x, y, overlap = FALSE)
# Report distance based on strand
x <- trbl_interval(~chrom, ~start, ~end, ~name, ~score, ~strand,"chr1", 10, 20, "a", 1, "-")
y <- trbl_interval(~chrom, ~start, ~end, ~name, ~score, ~strand,"chr1", 8, 9, "b", 1, "+","chr1", 21, 22, "b", 1, "-")
res <- bed_closest(x, y)
# convert distance based on strand
res$.dist_strand <- ifelse(res$strand.x == "+", res$.dist, -(res$.dist))
res
# report absolute distances
res$.abs_dist <- abs(res$.dist)
res
#######################################################################
## bed_clusterCluster neighboring intervals.
## 将有交集的bin归于一类
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 100, 200,
'chr1', 180, 250,
'chr1', 250, 500,
'chr1', 501, 1000,
'chr2', 1, 100,
'chr2', 150, 200)
bed_cluster(x)
# glyph illustrating clustering of overlapping and book-ended intervals
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 1, 10,
'chr1', 5, 20,
'chr1', 30, 40,
'chr1', 40, 50,
'chr1', 80, 90)
bed_glyph(bed_cluster(x), label ='.id')
#######################################################################
## bed_complementIdentify intervals in a genome not covered by a query.
## 取区间上没有交集的补集
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 0, 10,
'chr1', 75, 100)
genome <- trbl_genome(~chrom, ~size,'chr1', 200)
bed_glyph(bed_complement(x, genome))
genome <- trbl_genome(~chrom, ~size,
'chr1', 500,
'chr2', 600,
'chr3', 800)
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 100, 300,
'chr1', 200, 400,
'chr2', 0, 100,
'chr2', 200, 400,
'chr3', 500, 600)
# intervals not covered by x
bed_complement(x, genome)
#######################################################################
## bed_coverageCompute coverage of intervals.
## 计算两个bed文件之间交集的大小,以及在所占区间中的比例
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end, ~strand,
"chr1", 100, 500,'+',
"chr2", 200, 400,'+',
"chr2", 300, 500,'-',
"chr2", 800, 900,'-')
y <- trbl_interval(~chrom, ~start, ~end, ~value, ~strand,
"chr1", 150, 400, 100,'+',
"chr1", 500, 550, 100,'+',
"chr2", 230, 430, 200,'-',
"chr2", 350, 430, 300,'-')
bed_coverage(x, y)
#######################################################################
## bed_fisherFisher’s test to measure overlap between two sets of intervals.
#######################################################################
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
x <- bed_random(genome, seed = 1010486)
y <- bed_random(genome, seed = 9203911)
bed_fisher(x, y, genome)
#######################################################################
## bed_flankCreate flanking intervals from input intervals
## 取bed文件的两侧区间,常用来定义上下游区间
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 25, 50,
'chr1', 100, 125)
genome <- trbl_genome(~chrom, ~size,
'chr1', 130)
bed_glyph(bed_flank(x, genome, both = 20))
x <- trbl_interval(~chrom, ~start, ~end, ~name, ~score, ~strand,
'chr1', 500, 1000,'.','.','+','chr1', 1000, 1500,'.','.','-')
genome <- trbl_genome(~chrom, ~size,'chr1', 5000)
bed_flank(x, genome, left = 100) # 由于第一个区间[25,50]左边只有25bp,不满足100,这里是直接忽略此区间了,不与计算
bed_flank(x, genome, right = 100)
bed_flank(x, genome, both = 100)
bed_flank(x, genome, both = 0.5, fraction = TRUE) ## 表示上下游取该区间的50%范围
#######################################################################
## bed_glyphCreate example glyphs for valr functions.
## 此功能常用来对bed进行基因的注释
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 25, 50,
'chr1', 100, 125)
y <- trbl_interval(~chrom, ~start, ~end, ~value,
'chr1', 30, 75, 50)
bed_glyph(bed_intersect(x, y))
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 30, 75,
'chr1', 50, 90,
'chr1', 91, 120)
bed_glyph(bed_merge(x))
bed_glyph(bed_cluster(x), label ='.id')
#######################################################################
## bed_intersectIdentify intersecting intervals.
## 取y与x不相交的区间,
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 25, 50,
'chr1', 100, 125)
y <- trbl_interval(~chrom, ~start, ~end,
'chr1', 30, 75)
bed_glyph(bed_intersect(x, y))
bed_glyph(bed_intersect(x, y, invert = TRUE))
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 100, 500,
'chr2', 200, 400,
'chr2', 300, 500,
'chr2', 800, 900)
y <- trbl_interval(~chrom, ~start, ~end, ~value,
'chr1', 150, 400, 100,
'chr1', 500, 550, 100,
'chr2', 230, 430, 200,
'chr2', 350, 430, 300)
bed_intersect(x, y)
bed_intersect(x, y, invert = TRUE)
# start and end of each overlapping interval
res <- bed_intersect(x, y)
dplyr::mutate(res,
start = pmax(start.x, start.y),
end = pmin(end.x, end.y))
z <- trbl_interval(~chrom, ~start, ~end, ~value,
'chr1', 150, 400, 100,
'chr1', 500, 550, 100,
'chr2', 230, 430, 200,
'chr2', 750, 900, 400)
bed_intersect(x, y, z)
bed_intersect(x, exons = y, introns = z)
# a list of tbl_intervals can also be passed
bed_intersect(x, list(exons = y, introns = z))
#######################################################################
## bed_jaccard Calculate the Jaccard statistic for two sets of intervals
# 可以查看两个bed文件的交集的个数,独立的个数以及百分比,可以按照染色体分组
#######################################################################
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
x <- bed_random(genome, seed = 1010486)
y <- bed_random(genome, seed = 9203911)
bed_jaccard(x, y)
# calculate jaccard per chromosome
bed_jaccard(dplyr::group_by(x, chrom), dplyr::group_by(y, chrom))
# •len_i length of the intersection in base-pairs
# •len_u length of the union in base-pairs
# •jaccard value of jaccard statistic
# •n_int number of intersecting intervals betweenxandy
#######################################################################
## bed_makewindowsDivide intervals into new sub-intervals ("windows").
# 这个常用函数,将区间分成若干个bin,然后统计bin里面的数量
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end, ~name, ~score, ~strand,
"chr1", 100, 200,'A','.','+')
bed_glyph(bed_makewindows(x, num_win = 10), label ='.win_id')
# Fixed number of windows
bed_makewindows(x, num_win = 10)
# Fixed window size
bed_makewindows(x, win_size = 10)
# Fixed window size with overlaps
bed_makewindows(x, win_size = 10, step_size = 5)
# reverse win_id
bed_makewindows(x, win_size = 10, reverse = TRUE)
# bedtools'name num'
wins <- bed_makewindows(x, win_size = 10)
dplyr::mutate(wins, namenum = stringr::str_c(name,'_', .win_id))
#######################################################################
## bed_mapCalculate summaries from overlapping intervals.
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 100, 250,
'chr2', 250, 500)
y <- trbl_interval(~chrom, ~start, ~end, ~value,
'chr1', 100, 250, 10,
'chr1', 150, 250, 20,
'chr2', 250, 500, 500)
bed_glyph(bed_map(x, y, value = sum(value)), label ='value')
# summary examples
bed_map(x, y, .sum = sum(value))
bed_map(x, y, .min = min(value), .max = max(value))
# identify non-intersecting intervals to include in the result
res <- bed_map(x, y, .sum = sum(value))
x_not <- bed_intersect(x, y, invert = TRUE)
dplyr::bind_rows(res, x_not)
# create a list-column
bed_map(x, y, .values = list(value))
# use`nth`family from dplyr
bed_map(x, y, .first = dplyr::first(value)) # 可以用来统计第一个外显子之类的
bed_map(x, y, .absmax = abs(max(value)))
bed_map(x, y, .count = length(value)) # 可以统计区间内某目的元件的个数
bed_map(x, y, .vals = values(value))
# count defaults are NA not 0; differs from bedtools2 ...
bed_map(x, y, .counts = n()) # 功能类似length(value)
# ... but NA counts can be coverted to 0's
dplyr::mutate(bed_map(x, y, .counts = n()), .counts = ifelse(is.na(.counts), 0, .counts))
#######################################################################
## bed_mergeMerge overlapping intervals.
# 合并有交集的bin,计算bin里面基因的个数或者reads数量的总和
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 1, 50,
'chr1', 10, 75,
'chr1', 100, 120)
bed_glyph(bed_merge(x))
x <- trbl_interval(~chrom, ~start, ~end, ~value, ~strand,
"chr1", 1, 50, 1,'+',
"chr1", 100, 200, 2,'+',
"chr1", 150, 250, 3,'-',
"chr2", 1, 25, 4,'+',
"chr2", 200, 400, 5,'-',
"chr2", 400, 500, 6,'+',
"chr2", 450, 550, 7,'+')
bed_merge(x)
bed_merge(x, max_dist = 100)
# merge intervals on same strand
bed_merge(dplyr::group_by(x, strand))
bed_merge(x, .value = sum(value)) # 可以用来统计该区间内reads之类的总和
#######################################################################
## bed_partition Partition intervals into elemental intervals
# 将bin按照bed文件里面交集部分进行分割
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end, ~value, ~strand,
'chr1', 100, 500, 10, "+",
'chr1', 200, 400, 20, "-",
'chr1', 300, 550, 30, "+",
'chr1', 550, 575, 2, "+",
'chr1', 800, 900, 5, "+" )
bed_glyph(bed_partition(x))
bed_glyph(bed_partition(x, value = sum(value)), label = "value")
bed_partition(x)
# compute summary over each elemental interval
bed_partition(x, value = sum(value))
# partition and compute summaries based on group
x <- dplyr::group_by(x, strand)
bed_partition(x, value = sum(value))
# combine values across multiple tibbles
y <- trbl_interval(~chrom, ~start, ~end, ~value, ~strand,
'chr1', 10, 500, 100, "+",
'chr1', 250, 420, 200, "-",
'chr1', 350, 550, 300, "+",
'chr1', 550, 555, 20, "+",
'chr1', 800, 900, 50, "+" )
x <- dplyr::bind_rows(x, y)
bed_partition(x, value = sum(value))
#######################################################################
## bed_projectionProjection test for query interval overlap.
#######################################################################
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
x <- bed_random(genome, seed = 1010486)
y <- bed_random(genome, seed = 9203911)
bed_projection(x, y, genome)
bed_projection(x, y, genome, by_chrom = TRUE)
#######################################################################
## bed_random Generate randomly placed intervals on a genome.
# 随机产生长度为n的区间N个
#######################################################################
genome <- trbl_genome(~chrom, ~size,
"chr1", 10000000,
"chr2", 50000000,
"chr3", 60000000,
"chrX", 5000000)
bed_random(genome, seed = 10104)
# sorting can be suppressed
bed_random(genome, sort_by = NULL, seed = 10104)
# 500 random intervals of length 500
bed_random(genome, length = 500, n = 500, seed = 10104)
#######################################################################
## bed_reldist Compute relative distances between intervals.
#######################################################################
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
x <- bed_random(genome, seed = 1010486)
y <- bed_random(genome, seed = 9203911)
bed_reldist(x, y)
bed_reldist(x, y, detail = TRUE)
#######################################################################
## bed_shift Adjust intervals by a fixed size
# 将bed文件整体平移,正数表示向右平移,负数表示向左平移,也可以按照链方向来平移
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 25, 50,
'chr1', 100, 125)
genome <- trbl_genome(~chrom, ~size,
'chr1', 125)
bed_glyph(bed_shift(x, genome, size = -20))
x <- trbl_interval(~chrom, ~start, ~end, ~strand,
"chr1", 100, 150, "+",
"chr1", 200, 250, "+",
"chr2", 300, 350, "+",
"chr2", 400, 450, "-",
"chr3", 500, 550, "-",
"chr3", 600, 650, "-")
genome <- trbl_genome(~chrom, ~size,
"chr1", 1000,
"chr2", 2000,
"chr3", 3000)
bed_shift(x, genome, 100)
bed_shift(x, genome, fraction = 0.5)# shift with respect to strand
stranded <- dplyr::group_by(x, strand)
bed_shift(stranded, genome, 100)
#######################################################################
## bed_shuffle Shuffle input intervals
#######################################################################
genome <- trbl_genome(~chrom, ~size,
"chr1", 1e6,
"chr2", 2e6,
"chr3", 4e6)
x <- bed_random(genome, seed = 1010486)
bed_shuffle(x, genome, seed = 9830491)
#######################################################################
## bed_slopIncrease the size of input intervals
# 扩展区域空间,延长区间上下游
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 110, 120,
'chr1', 225, 235)
genome <- trbl_genome(~chrom, ~size,
'chr1', 400)
bed_glyph(bed_slop(x, genome, both = 20, trim = TRUE))
genome <- trbl_genome(~chrom, ~size,
"chr1", 5000)
x <- trbl_interval(~chrom, ~start, ~end, ~name, ~score, ~strand,
"chr1", 500, 1000,'.','.','+',
"chr1", 1000, 1500,'.','.','-')
bed_slop(x, genome, left = 100)
bed_slop(x, genome, right = 100)
bed_slop(x, genome, both = 100)
bed_slop(x, genome, both = 0.5, fraction = TRUE)
#######################################################################
## bed_sort Sort a set of intervals
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
"chr8", 500, 1000,
"chr8", 1000, 5000,
"chr8", 100, 200,
"chr1", 100, 300,
"chr1", 100, 200)
# sort by chrom and start
bed_sort(x)
# reverse sort order
bed_sort(x, reverse = TRUE)
# sort by interval size
bed_sort(x, by_size = TRUE)
# sort by decreasing interval size
bed_sort(x, by_size = TRUE, reverse = TRUE)
# sort by interval size within chrom
bed_sort(x, by_size = TRUE, by_chrom = TRUE)
#######################################################################
## bed_subtract Subtract two sets of intervals.
# 提取a.bed文件中不包含b.bed文件的区间, any参数表示a在b中一点交集也没有的区间
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 1, 100)
y <- trbl_interval(~chrom, ~start, ~end,
'chr1', 50, 75)
bed_glyph(bed_subtract(x, y))
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 100, 200,
'chr1', 250, 400,
'chr1', 500, 600,
'chr1', 1000, 1200,
'chr1', 1300, 1500)
y <- trbl_interval(~chrom, ~start, ~end,
'chr1', 150, 175,
'chr1', 510, 525,
'chr1', 550, 575,
'chr1', 900, 1050,
'chr1', 1150, 1250,
'chr1', 1299, 1501)
bed_subtract(x, y)
bed_subtract(x, y, any = TRUE)
#######################################################################
## bed_window Identify intervals within a specified distance.
# 相当于将a.bed区间延长两端都延长 bp, 然后查找能与b相交的a中的区间
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 25, 50,
'chr1', 100, 125)
y <- trbl_interval(~chrom, ~start, ~end,
'chr1', 60, 75)
genome <- trbl_genome(~chrom, ~size,
'chr1', 125)
bed_glyph(bed_window(x, y, genome, both = 15))
x <- trbl_interval(~chrom, ~start, ~end,
"chr1", 10, 100,
"chr2", 200, 400,
"chr2", 300, 500,
"chr2", 800, 900)
y <- trbl_interval(~chrom, ~start, ~end,
"chr1", 150, 400,
"chr2", 230, 430,
"chr2", 350, 430)
genome <- trbl_genome(~chrom, ~size,
"chr1", 500,
"chr2", 1000)
bed_window(x, y, genome, both = 100)
#######################################################################
## bound_intervals Select intervals bounded by a genome.
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
"chr1", -100, 500,
"chr1", 100, 1e9,
"chr1", 500, 1000)
genome <- read_genome(valr_example('hg19.chrom.sizes.gz'))
# out-of-bounds are removed by default ...
bound_intervals(x, genome)
# ... or can be trimmed within the bounds of a genome
bound_intervals(x, genome, trim = TRUE)
#######################################################################
## create_introns Create intron features.
## 更具exon坐标生成intro坐标
#######################################################################
x <- read_bed12(valr_example('mm9.refGene.bed.gz'))
create_introns(x)
#######################################################################
## create_tss Create transcription start site features.
# 提取转录终止位点
#######################################################################
x <- read_bed12(valr_example('mm9.refGene.bed.gz'))
create_tss(x)
#######################################################################
## create_utrs3 Create 3’ UTR features.
## 提取3' UTR区间
#######################################################################
x <- read_bed12(valr_example('mm9.refGene.bed.gz'))
create_utrs3(x)
#######################################################################
## create_utrs5 Create 5’ UTR features.
#######################################################################
x <- read_bed12(valr_example('mm9.refGene.bed.gz'))
create_utrs5(x)
#######################################################################
## dbFetch data from remote databases
#######################################################################
## Not run:
if(require(RMySQL)) {
ucsc <- db_ucsc('hg38')
# fetch the`refGene`tbl
tbl(ucsc, "refGene")
# the`chromInfo`tbls have size information
tbl(ucsc, "chromInfo")
}
## End(Not run)
## Not run:
if(require(RMySQL)) {
# squirrel genome
ensembl <- db_ensembl('spermophilus_tridecemlineatus_core_67_2')
tbl(ensembl, "gene")
}
#######################################################################
## flip_strandsFlip strands in intervals.
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end, ~strand,
'chr1', 1, 100,'+',
'chr2', 1, 100,'-')
flip_strands(x)
#######################################################################
## interval_spacingCalculate interval spacing.
# 表示第二个个bed区间的开始位点离第一个bed区间的终止位点的距离
#######################################################################
x <- trbl_interval(~chrom, ~start, ~end,
'chr1', 1, 150,
'chr1', 160, 200,
'chr2', 200, 300)
interval_spacing(x)
#######################################################################
## is.tbl_genome Test if the object is a tbl_genome.
#######################################################################
is.tbl_interval(x)
is.tbl_genome(x)
#######################################################################
## read_bed Read BED and related files
#######################################################################
# read_bed assumes 3 field BED format.
read_bed(valr_example('3fields.bed.gz'))
read_bed(valr_example('6fields.bed.gz'), n_fields = 6)
# result is sorted by chrom and start unless`sort = FALSE`
read_bed(valr_example('3fields.bed.gz'), sort = FALSE)
read_bed12(valr_example('mm9.refGene.bed.gz'))
read_bedgraph(valr_example('test.bg.gz'))
read_narrowpeak(valr_example('sample.narrowPeak.gz'))
read_broadpeak(valr_example('sample.broadPeak.gz'))
#######################################################################
## read_genome Read genome files
#######################################################################
read_genome(valr_example('hg19.chrom.sizes.gz'))
#`read_genome`accepts a URL
read_genome('https://genome.ucsc.edu/goldenpath/help/hg19.chrom.sizes')
#######################################################################
## read_vcf Read a VCF file.
######################################################################
vcf_file <- valr_example('test.vcf.gz')
read_vcf(vcf_file)
#######################################################################
## tbl_genomeTibble for reference sizes.
######################################################################
genome <- tibble::tribble(~chrom, ~size,'chr1', 1e6,'chr2', 1e7)
is.tbl_genome(genome)
genome <- tbl_genome(genome)
is.tbl_genome(genome)
trbl_genome(~chrom, ~size,
'chr1', 1e6)
#######################################################################
## tbl_interval Tibble for intervals.
######################################################################
x <- tibble::tribble(~chrom, ~start, ~end,
'chr1', 1, 50,
'chr1', 10, 75,
'chr1', 100, 120)
is.tbl_interval(x)
x <- tbl_interval(x)
is.tbl_interval(x)
#######################################################################
## valr valr: genome interval arithmetic in R
######################################################################
valr_example('hg19.chrom.sizes.gz')