之前在上看过一篇文章https://www.jianshu.com/p/1f5d13cc47f8是重复16年发表在NATURE PROTOCOLS上面的一篇处理RNA-seq数据的文章,文章名字是:Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown,主要讲了用Hisat2、StringTie、Ballgown三款软件对转录组的数据进行分析,这三款软件的分析效率比较高,今天我尝试用大家熟悉的老套路老方法分析下这组数据。
文章中的所有数据如下图,我是用win10中wsl系统进行分析的(无奈,没有服务器啊!),在这就不做质控了,直接从建立索引开始。
我的是Ubuntu系统,安装bowtie2直接命令行
$ sudo apt install bowtie2
#bowtie2建立chrX索引
$ bowtie2-build chrX.fa chrX

然后用tophat2进行比对,为了节省时间,写了个脚本1.sh:
for i in {188044,188144,188145,188257,188273,188337,188383,188401,188428,188454,
204916};do tophat2 –p 4 –o ERR${i}-out /mnt/f/data/chrX_data/genome/chrX ./samples /ERR${i}_chrX_1.fastq.gz ./ samples /ERR${i}_chrX_2.fastq.gz
done
但是在执行时却出现了报错,具体报错信息贴在下面了:
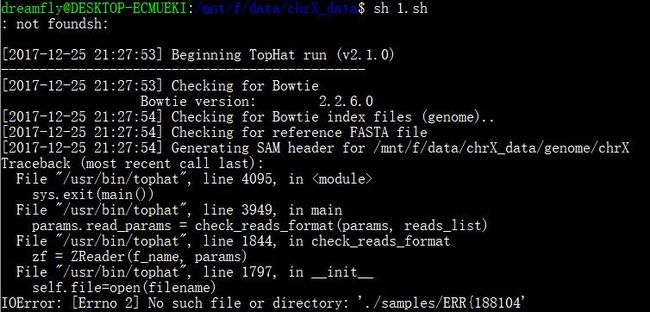
哪位大神能够给指导下,在线等·······································

而每个输出文件的内容大概有以下6个文件和一个日志文件夹,以输出的ERR188044为例:

接下来分析我们主要用到accepted_hits.bam文件,利用cuffdiff基于参考注释对这12个样本进行表达差异分析,具体代码如下:
cuffdiff -o expdiff –b ./genome/chrX.fa –L ERR188044, ERR188104, ERR188234, ERR188245, ERR188257, ERR188273, ERR188337, ERR188383, ERR188401, ERR188428,ERR188454,ERR204916 -u ./genes/chrX.gtf ./ERR188044-out/accepted_hits.bam ./ERR188104-out/accepted_hits.bam ./ERR188234-out/accepted_hits.bam ./ERR188245-out/accepted_hits.bam ./ERR188257-out/accepted_hits.bam ./ERR188273-out/accepted_hits.bam ./ERR188337-out/accepted_hits.bam ./ERR188383-out/accepted_hits.bam ./ERR188401-out/accepted_hits.bam ./ERR188428-out/accepted_hits.bam ./ERR188454-out/accepted_hits.bam ./ERR204916-out/accepted_hits.bam

接下来就利用R语言中cummeRbund包对结果进行可视化分析了,你们自由发挥吧。
请关注我的公众号----生信栈,不定期分享实用的生物信息干货!!!
