Chromosomal-level reference genome of Chinese peacock butterfly (Papilio bianor) based on third-generation DNA sequencing and Hi-C analysis

Abstract
来自昆明动物所的研究人员通过pacbio与Hi-c技术的结合从头测序得到了碧凤蝶染色体级别的基因组。最终组装的基因组大小是421.52Mb,30条染色体(29 autosome and 1 Z sex chromosome),scaffoldN50为13.12Mb,15,375个蛋白编码基因以及233.09Mb的重复序列,物种进化分歧分析表明碧凤蝶和柑橘凤蝶有共同的祖先,两个物种的分歧时间大概在23.69-36.07百万年前。种群历史数量统计表明,物种的种群扩增发生在最后一次间冰期到最后一次盛冰期之间,这可能是由于天敌以及对冰期气候环境的适应。
这篇文章的原始数据:https://www.ebi.ac.uk/ena/data/view/PRJNA530186
分析过程软件参数及代码:ftp://parrot.genomics.cn/gigadb/pub/10.5524/100001_101000/100653/
代码网盘链接地址:链接:https://share.weiyun.com/5e6tovj 密码:hktmze
Backgroud
蝴蝶是一种在全世界范围内非常具有观赏性,吸引性的动物,由于其不同寻常的翅纹在各个物种,性别以及季节都有不同的表现,尤其是拟态理论的提出,让蝴蝶再次成为了焦点。但由于昆虫基因组的高杂合度,目前只有6个科,37种蝴蝶有他们的参考基因组,而染色体级别的基因组只有红带袖蝶,庆网蛱蝶以及柑橘凤蝶,这些染色体都是通过构建遗传图谱得到的。
随着第三代单分子测序技术的发展,再结合高通量染色体构象捕获技术,可以得到染色体的图谱,目前已经应用到果蝇,蚊子以及蛾子中。
Female adult. Left, dorsal view; right, ventral view
Data Description
Insect collection and breeding
2个五龄幼虫用于Hi-C测序,一头雄蛾用于genome survey以及de novo genome sequencing。
Genome survey using Illumina sequencing technology
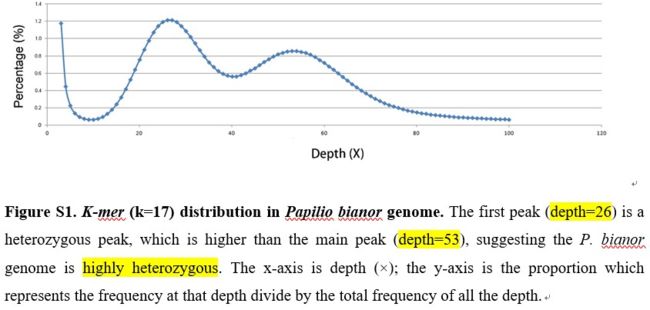
一头雄蛾的胸和腹用于提取基因组DNA,Illumina HiSeq2000构建150和500bp两种插入片段大小的文库,kmer=17,G =
k-mer number/k-mer depth,预估的基因组大小为496.05 Mb,杂合度1.81%。
Library construction and sequencing using SMRT and Hi-C technologies
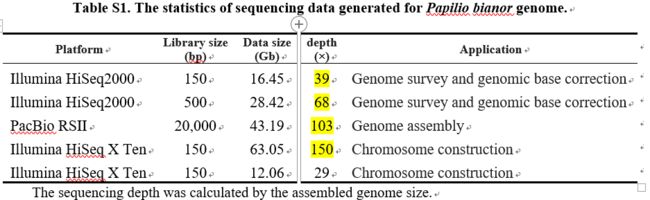
提取基因组DNA后,构建20kb文库用于Pacbio测序,10个SMRT cells( PacBio RSII platform)用于测序,共产生43.19 Gb subreads,平均长度16.4kb,400–700 bp的文库( Illumina
HiSeq X Ten platform), PE150测序,产生∼75.11 Gb
raw reads。
Chromosomal-level genome assembly
考虑到碧凤蝶基因组的高杂合度。First,PacBio-only assembly using Wtdbg(–tidyreads 5000 -k 0 -p 17 -S 1),Wtdbg是一款基于fuzzy Bruijn graph算法的组装软件,用于组装PacBio以及nanapore的测序数据。Second,用Illumina reads去polish PacBio-only assembly sequences:先将Illumina reads mapping到PacBio-only assembly sequences,再用Pilon做两轮的碱基矫正。Third,由于矫正后的基因组仍有很多低测序深度的short contigs,将identity>90%,低测序深度的短contigs(size < 1,000 bp and coverage < 50 or size < 10,000 bp and coverage < 35)merge成更长的contigs。Fourth,用Juicer软件和3D de novo assembly将Hi-C的raw reads比对到polished assembly genome上去提高组装质量。
最终90.5.%的contigs被固定到30个uper-scaffolds上,这可能对应着30条染色体。最后,得到了碧凤蝶染色体级别的基因组,基因组大小是421.52Mb,scaffoldN50为13.12Mb,大约占预估基因组大小的85%(高杂合的表现)。
Heat map of chromosomal interactions. Each chromosome is framed with a blue block, and each scaffold is framed with a green block.
Quality evaluation of assembled genome
基因组组装质量的评估主要通过三个方法。3C原则(completeness, base level contiguity, and accuracy)。First,基因组的完整性评估通过BUSCO评价(insecta_odb9),core genes的覆盖度为96.60%。Second,通过BWA和BLASER计算Illumina and PacBio reads与组装基因组的mapping rates,最终96.31% of Illumina reads mapped to the assembled genome with few heterozygous regions
96.86% of PacBio reads also mapped to the assembled genome
with few heterozygous regions。Third,通过碧凤蝶基因组与柑橘凤蝶基因组染色体共线性分析,61,082,412 bp of the P. bianor assembled genome could be aligned (1:1) with high confidence
(-m 0.01) to the P. xuthus reference genome。

Circos plot of P. bianor chromosome-level reference genome with the previously released Papilio xuthus genome (obtained from a Chinese group). Shown from outermost to innermost are (1) gene density, (2) repeat element density, (3) GC content, and (4) syntenic regions with P. xuthus (left).
Genome annotation
基因组注释:
- 重复序列注释
- 基因结构注释
- 基因功能注释
- 重复序列注释
重复序列包括串联重复序列以及散在重复序列。
串联重复序列主要包括微卫星,小卫星等短的串联重复。
散在重复序列包括DNA/RNA转座子,LTR(long terminal repeats),LINE(long interspersed nuclear elements),SINE(short interspersed nuclear elements)等。
总的来讲,基因组的注释主要通过基于从头预测以及基于同源比对的方法。
重复序列
Tandem Repeats Finder to annotate the tandem repeats(Tandem Repeats Database)
-
TEs de novo and homology-based approaches at both the DNA and protein levels
-
At the DNA level
- RepeatModeler to construct a de novo repeat library
- Repeat-Masker to search similar TEs against the known Repbase TE library and de novo repeat library
- LTR FINDER to find long terminal repeats (LTRs)
-
At the protein level
- RepeatProteinMask search the assembled genome against the TE protein database using the WU-BLASTX engine
-
蛋白编码基因
-
结构注释
- de novo gene prediction: the repeat-masked genome was analyzed by SNAP GENSCAN glimmerHMM AUGUSTUS
- homology-based predictions: TBLASTN with an E-value cut-off of 1e−5 to align the protein sequences of the reference gene set to the genome, and GeneWise to perform more precise alignment
- Evidence-Modeler software was used to integrate the genes predicted by the homology and de novo approaches and generate a comprehensive, non-redundant gene set
-
功能注释
- KEGG, TrEMBL, SwissProt, and COG databases were searched for best matches to P. bianor for the protein sequences yielded by EVM software, using BLASTP
- Pfam, PRINTS, ProDom, and SMART databases were searched for known motifs and domains in our sequences using Inter-ProScan software
-
searched all predicted gene sequences against the GenBank nonredundant protein (nr) database using BLASTP
 transposable elements (TEs)
transposable elements (TEs)
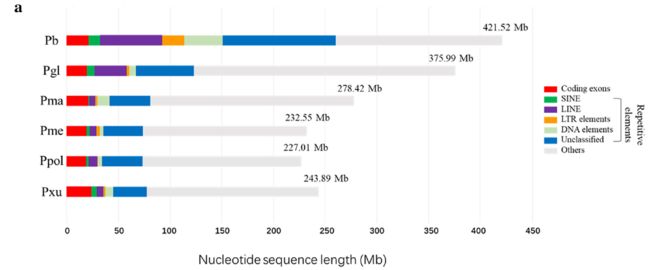 图片.png
图片.png
(a) Breakdown of the whole-genome assemblies into different functional classes in Papilio
基因家族鉴定与进化分析
-
OrthoMCL to cluster the annotated genes
 图片.png
图片.png
(b) Venn diagram of the shared gene families of Papilio.The result showed that 293 gene families were specific to P. bianor.
Using CAFE identified expanded gene families and contracted gene families
A total of 1,378 one-to-one single-copy orthologs that contain
only 1 protein for each species were collected and clustered
by OrthoMCL-
nucleic acid sequences were aligned using PRANK,Gene alignments were concatenated and phylogenetic trees were constructed using RAxML with GTR+G+I model
 图片.png
图片.png -
the phylogeny was further analyzed by MCMCtree in PAML to investigate the divergence time of these species
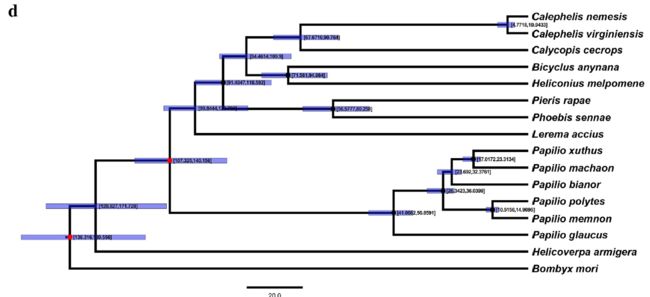 图片.png
图片.png
(d) Maximum likelihood phylogenetic tree of Papilionoidea constructed by the concatenated alignment of 1,378 1-to-1 single-copy ortholog genes. The numbers in the square brackets on the nodes are the 95% confidence intervals of divergence time. The red dots are fossil evidence downloaded from the TimeTree website , and the black dots are inferred time obtained from the TimeTree website. Both were used to calibrate divergent time.
-
demographic histories applying the Pairwise Sequentially Markovian Coalescence analysis mapping Illumina short reads to the assembled genome with BWA and calling variants with SAMtools
 图片.png
图片.png
(c) The dynamic changes of the effective population size were plotted using PSMC software, with 100 bootstrap replicates to test the robust variations. The parameter “g” represents the generation time in years, and the parameter “μ” means the per generation mutation rate.
下期预告:

https://www.nature.com/articles/s41467-019-12175-9
reference:
https://academic.oup.com/gigascience/article/8/11/giz128/5612101