计算机辅助药物设计的主要包括两种方法:1、基于受体的药物设计。2、基于配体的药物设计。当蛋白的晶体结构以及结合位点已知时,可以使用基于受体的药物设计方法,例如分子对接。然而分子对接方法存在缺陷,例如:无法正确的处理诱导契合然效应、溶剂化效应、打分函数的排序能力较差等。此外由于大量蛋白的晶体结构仍然是未知的,尤其是膜蛋白, 膜蛋白极其疏水的特性使其纯化结晶变得很困难。对晶体结构未知的靶点,当存在多种的结构类似的配体时,可以使用基于药效团的药物设计方法。

在成药靶点中必定存在着能与药物结合的特异性结合位点。对某个靶点发挥活性的化合物在结构特征上必定有相似之处。这些化合物的最普遍的共有特性被定义为药效团(pharmacophore)。IUPAC将药效团定义为“确保与特定生物靶标的最佳相互作用并触发其生物反应”所必需的空间和电子特征的集合。
药效团模型的重要意义:
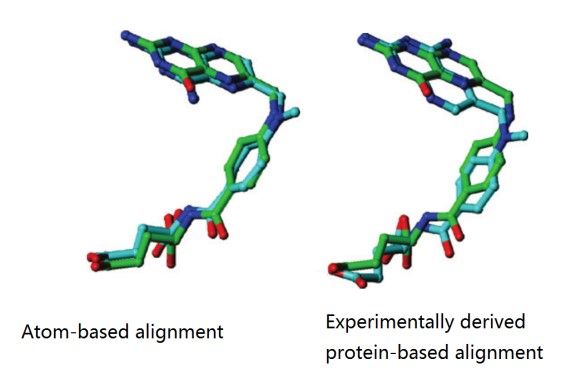
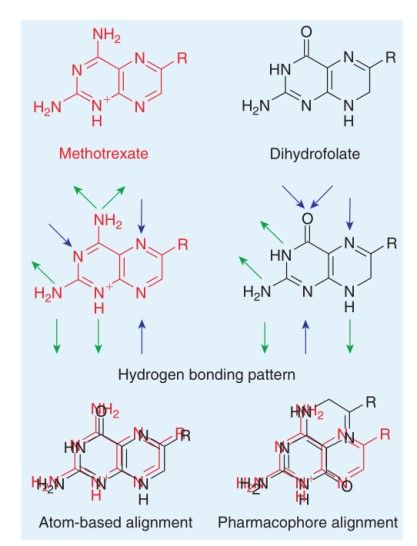
药效团模型不仅仅利用分子拓扑学相似性而且利用了基团的功能相似性,从而运用了生物电子等排体(bioisosterism)的概念使得模型更加可靠。图2比较了甲氨蝶呤和二氢叶酸两个化合物基于拓扑学相似性的叠合以及实验验证的构象叠合(1rx2,1rb3),指出基于药效团的构象与实验结果更加接近。如果仅仅考虑化合物之间形状的相似性,将会导致结合模式预测错误。如果将分子的药效团特征(氢键受体、氢键供体考虑在内)则会纠正这一错误。
基于药效团的虚拟筛选的方法是可靠而且快速的虚拟筛选工具。基于受体的虚拟筛选方法对百万级别的化合物库而言筛选速度缓慢。除了速度这一因素,基于受体的虚拟筛选方法很少考虑蛋白柔性、水分子的影响、溶剂化效应、以及配体的构象限制。尽管基于受体的虚拟筛选方法可以提供配体与蛋白的相互作用信息,但是精确地预测蛋白与小分子的结合力仍然是一个无法解决的问题。
药效团的应用:
在计算机辅助药物设计中,药效团模型主要被用于三个领域:
1、发现药物分子的关键药效特征从而建立清晰的构效关系(structure activity relationship);
2、骨架跃迁(scaffoldhopping),通过药效团的方法对化合物库进行虚拟筛选发现对靶点有活性的新骨架化合物;
3、通过药效团进行靶点预测(targetfishing),预测化合物的药理学作用谱,通过使用药效团模型,有可能在药物开发的早期预测先导的不良反应,从而减少药物研发失败的概率。

药效团模型构建方法:
为了获得准确的药效团模型,首先必须使用正确的化合物的3D结构。因此原子价、键级、质子化状态、互变异构以及立体异构等因素都必须要进行仔细的检查。此外获得准确的药效团模型的另一个前提是,用于构建模型的化合物具有相似的结合模式。
构建药效团的流程为:
1、挑选一组对特定靶点中相同结合位点有活性的配体。
2、对所有配体进行构象分析。
3、指定药效团特征。
4、对配体构象进行叠合从而获得药效团模型。
配体的构象分析以及生物活性构象:
由于化合物是柔性和动态的,因此,必须使用构象分析的方法生成低能构象系综。构象分析在药效团模型构建过程中是至关重要的步骤,这是由于构象分析的目的不仅仅是获得化合物的全局能量最小化构象,还有获得化合物的生物活性构象。
为了与一个受体结合且具有较高的亲和力,配体必须与靶点的结合口袋相匹配。静态匹配主要依赖于配体的构象。在结合口袋内,配体的构象未必总是能量最低构象,这是由于配体从受体中获得相互作用能,将会抵消高能构象的影响。高能构象对自由结合能的影响如公式所示:

如果化合物的结合构象与能量最小化结构的能量偏差达到1.42kcal/mol,则化合物的亲和力将会减少一个数量级。因此具有高亲和力的化合物的结合构象一般都是能量较小的。目前大量的构象分析方法已经被开发。普遍使用的构象方法如下所示。
系统搜索方法:
系统搜索方法通过逐渐旋转每一个可旋转键来获得最优构象。系统搜索方法的优点在于,构象搜索较为全面。然而在可旋转键较多化合物中,该种方法的计算成本很高。
随机搜索方法:
最常使用的随机搜索方法为蒙特卡罗(Monte Carlo)方法,该方法通过随机旋转可旋转键(旋转角度为为一个随机数)。该方法起始于一个能量最小化的构象A,随后对构象A进行随机的构象搜索获得构象B。当Epot (B) < Epot (A)时新的构象B被接纳为能量最小化构象。蒙特卡罗方法能有效的对构象空间进行采样,然而和所有的随机搜索工具一样,不能保证所有的构象搜索的完整性。另一个有效地随机筛选方法是poling法。该方法已植入Catalyst程序中。
模拟退火方法:
模拟退火算法是基于分子动力学的构象搜索方法,该方法假定分子中的原子会按照力场的规则进行相互作用。在模拟退火方法中,系统的温度突然升高使得分子具有足够的内能克服能量壁垒。随后系统被缓慢冷却从而获得能量合适的构象。模拟退火方法的计算成本较高,因此仅仅使用在分子集较小的情况下。和蒙特卡洛方法一样,该方法不能保证所有构象搜索的完整性。
多个研究发现多个构象搜索工具均能产生的低能构象系综中包括了实验所观察到的活性构象。哪一种构象搜索工具的功能最为强大?这个问题的答案取决于研究的数据集和需要解决的问题。如果数据集化合物的数量较少则使用计算成本较高的计算方法,例如系统搜索方法。如果数据集中的化合物数量较多,则应该使用更为快速和简化的方法,例如Catalyst或者Omega。
分子叠合技术:
目前已有多种分子叠合技术,例如通过比较化合物结构的力场性质以及形状性质为基础的方法(DISCO、Catalyst、LigandScout)。另一种构象叠合方法是是FlexS算法。当应用FlexS算法时,柔性化合物首先被拆分为片段,随后锚定片段以与参考分子相互作用相类似的的方式进行叠合,然后柔性分子的剩余片段被逐渐加入。分子的柔性得到充分考虑以保证叠合构象是能量较低的构象。
指定药效团特征:
组胺H3受体拮抗剂的药效团模型如下图所示。其中包含了6个关键的药效团元素,分别是正电基团、氢键供体基团、氢键受体基团、脂肪族疏水基团、芳香基团,疏水作用基团等。除此以外,分子形状可以被合并到药效团特征中。


分子匹配药效团的难易程度,由药效团模型中定义的药效特征数量以及药效团模型的容忍性决定。此外,分子能否匹配药效团还与分子的构象有关。当用于构建药效团模型的化合物的构象改变后,该分子可能无法匹配该药效团。因此,为了增加分子匹配药效团模型的可能性,该分子需要生成一组构象系综。定义实用的药效团模型的困难在于,药效团模型只包括与靶点结合必须的关键药效团元素。
参考文献:Chapter 28 Pharmacophore Identification and Pseudo-Receptor Modeling . InThe Practice of Medicinal Chemistry, 3rd Ed, 2008.