差异基因分析
做基因表达分析时必然要做差异基因分析,做差异基因分析最常用的软件就是DESeq2,使用DESeq2对两组进行比较,得到显著性P-value,P-adjust值和基因间的差异倍数(Fold Change)。根据这三个统计学参数来筛选差异基因,一般设置的筛选条件为:p-value<0.05和Fold change≥2及Fold change< 0.01。实际上会根据具体分析情况来调整参数的范围。
经过筛选,就得到了“xxx个差异基因”及其他详细内容,见下图
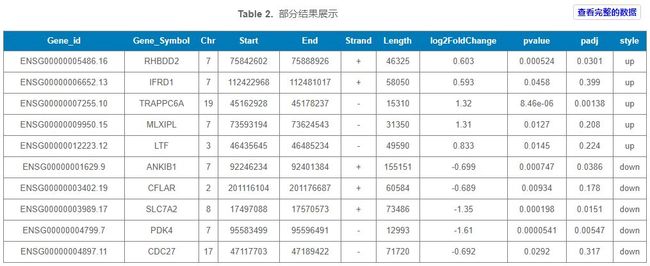
上图列头及其含义说明:
Gene_id:基因在Eesembl数据库的编号
Gene_Symbol:基因名称,与NCBI GenBank的基因标识一致
Chr:该基因所在染色体名称
Start:该基因在染色体上的起始位点
End:该基因在染色体上的终止位点
Strand:该基因所在染色体所属的链的属性。'+'表示正链,'-'表示负链
Length:该基因的长度
log2FoldChange:log2的差异倍数。本次取值大于1.5倍或者小于0.666667倍。
p-value:p值,用来表示差异基因的显著性水平,本次取p值小于0.05。
padj:p 值校正值 FDR,即对相同条件下的试验的差异显著性进行多重检验校正。
前面几个列头代表的意思很容易理解,我们最关注的是log2FoldChange、P值和Padjust值
P-value:p值,是统计学检验变量,代表差异显著性,一般认为P < 0.05 为显著, P <0.01 为非常显著。其含义为:由抽样误差导致样本间差异的概率小于0.05 或0.01。
Padj:p adjust。转录组测序的差异表达分析是对大量的基因表达值进行的独立统计假设检验,存在假阳性问题,因此引入Padj对显著性P值(P adjust)进行校正。Padj是对P-value的再判断,筛选更为严格。
Fold Change:适用于两分组分析,表示两样本(组)间表达量的比值。
log2FoldChange:对Fold Change取log2,一般默认表达相差2倍以上是有意义的,可以根据情况适当放宽至1.5/1.2,但最好不要低于1.2倍。

▲FC的计算方式,有兴趣可以详细看。
用上面的方法筛选到差异基因之后, 得到了非常多的数据。直接查看数据不方便观察整体情况,我们一般会将数据做成火山图来展示组间差异的整体情况。
火山图
火山图从差异倍数(Fold Change)和差异显著性水平(p-value)两个方向对组间差异进行评估,如图:
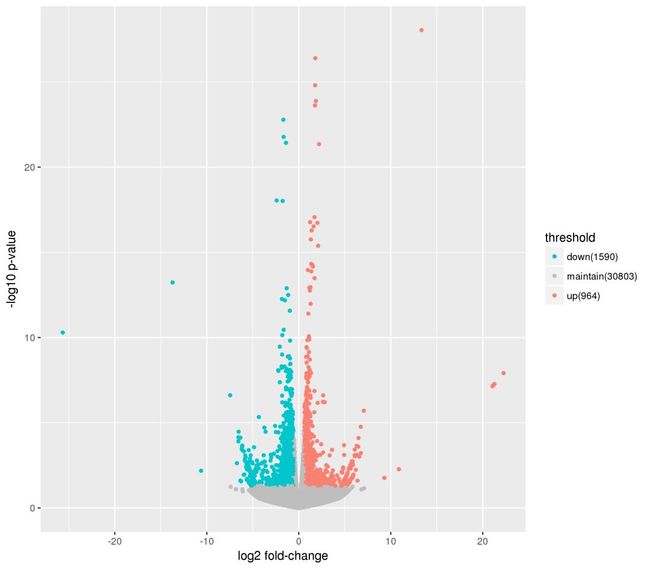
横轴:log2FoldChange,对fold change值取log2。横坐标的绝对值越大,说明表达量在两样本间的表达量倍数差异越大。
纵轴:-log10P-value,对P-vallue取-log10。纵坐标值越大,说明差异基因表达越显著,筛选得到的差异表达基因越可靠。
图中差异倍数大于1.5且p值小于0.05的为上调基因, 用红色点展示;差异倍数小于0.666667并且p值小于0.05的为下调基因, 用绿色点展示;其中非显著差异的基因用灰色点展示。
上面,我们提到了上调基因、下调基因。那么,什么是上调基因,下调基因?简单解释一下:
上调基因:up-regulated gene,相对于对照组,在实验组中该基因转录成mRNA时受到正向调控,表达量增加。
下调基因:down-regulatedgene,相对于对照组,在实验组中该基因转录成mRNA时受到抑制,表达量减少。
差异基因聚类分析
基因的作用不是孤立的,因此对差异基因进行层次聚类分析,从聚类分析结果中发现样本组内和组间的相关性情况。
(顾名思义,聚类分析可以基于分析对象的相似性将其各自聚成不同的组。)
根据每个基因在样本中的信号值来分别对基因和样本进行聚类分析,聚为一类的基因群在样本中具有相似的表达情况,这说明它们可能具有相似的功能。
聚类结果用heatmap(热图)进行展示:
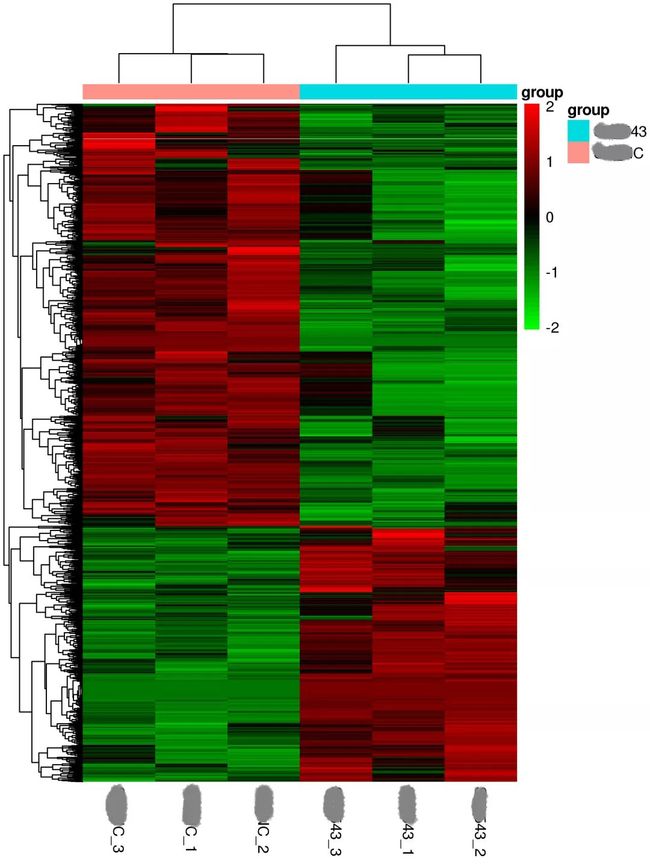
横坐标:样本名称
纵坐标:差异基因
从图上可以看出,样本分为“xxxC”和“xxx43”两组,红色代表该差异基因在分组样本中表达值高,绿色代表差异基因在分组样本中表达值低。不同列代表不同的样本。
来源:小L GCBI知识库,如有侵权可联系删除!