上次内容主要是介绍该review paper一些背景还有TE的分类,现在继续解读一下,下一部分的内容。
TE的探测与注释的方法
TE的探测和注释,可以在有或没有基因组assembly的情况下执行,这一部中常用到的工具如下图。
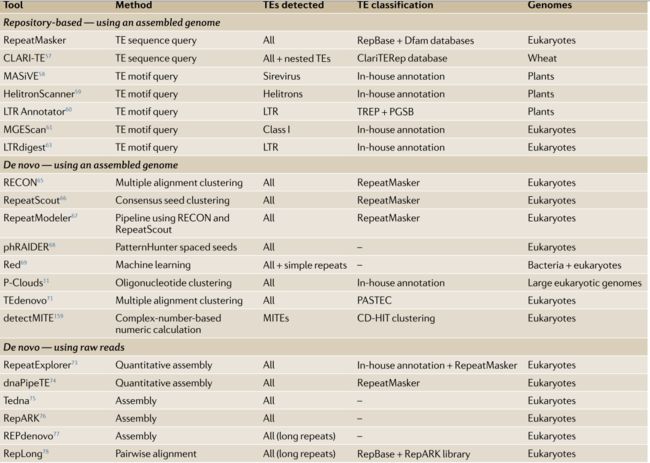
两种主要策略依赖于组装的基因组:首先是基于已有的数据库来的注释,其中根据已知的TE共有序列或TE基序查询序列;第二个是de novo注释。不需要基因组装配的替代方法是使用原始reads的从头注释。虽然基于存储库的RepeatMasker目前是TE注释的黄金标准,但是de novo方法提供了识别新TE系列的潜在性。这些方法可以作为基于存储库的搜索的第一步,以提供更全面的结果,这一功能已经包含在一些de novo注释工具中,用于TE注释与探索
基于库的注释
基于存储库的注释工具是对TE共有序列或与不同TE家族相关联的TE基序列进行全基因组搜索。其效果与所用数据库的质量和特异性有关。最广为人知的TE序列数据库查询工具是RepeatMasker,它针对RepBase Update和Dfam数据库进行查询,并为广泛的真核生物提供了散布重复序列和低复杂度DNA的公开列表。该工具目前将48%的人类基因组注释为TEs,当与Dfam2.0结合一起使用时,该百分比增加至53%。
RepeatMasker可以将重复序列标记为多个N。
这一步成为绕过与重复序列相关的分析问题的有效策略,但于此同时也会“掩盖”重复的下游分析。对于特定基因组或TE家族也存在其他工具。这类软件包括CLARI-T,一款最近开发用于注释植物的复杂基因组,包括在小麦基因组中注释频繁的嵌套重复序列。此外的其他工具搜索也可以用于搜索特定TE家族相关的基序或基因组结构。它们包括MASiVE,HelitronScanner和LTR Annotator,等,可以检测植物基因组中的Helitrons和LTRs。
基于基因组的重头注释
在过去,一些最流行的用于组装基因组的de novo注释工具是RECON,RepeatScout和RepeatModeler,它们使用成对相似性或共有种子作为聚类重复序列的起始点。基于与RepeatScout相同的原理,据报道,最近的phRAIDER工具速度提高了十倍,可以识别TE和简单的重复。这些经典的工具一直都是应用于注释新测序的基因组,以及从RepeatMasker基因组注释中识别缺失的TE。实际上,使用这些从头方法的人类基因组注释通常会导致额外的10%或更多被分配给TE。这可能包括误报假阳性的TE,但也包括新的TE家族和TE实例,这些实例与其家族共识不同,无法识别,所以会多了出来。
使用raw reads进行重头注释
与其他基因组序列相比,该物种中的工具利用高丰度的TE序列,并使用低覆盖度测序数据直接从原始读数组装TE和其他重复序列。从这个趋势开始,RepeatExplorer最初作为基于图形的聚类算法发布,用于从454测序reads中鉴定TE,然后使用RepeatMasker对每个簇进行注释。已发布更高版本的RepeatExplorer,具有扩展功能并部署在Galaxy服务器上。遵循相同的原则,dnaPipeTE使用Trinity汇编程序开发用于下一代测序(NGS)short reads。与RepeatExplorer类似,dnaPipeTE生成TE的定量注释,但也通过reads重叠群比较提供TE的ages。其他此类工具包括Tedna和RepARK;但是,这两个工具缺乏
TE的基因组定量。最后,REPdenovo也从原始reads中汇集了TE,其开发部分是为了解决长重复序列的组装问题,并确定了人类基因组中缺失的参考TE实例。总之,这些TE组装工具代表了在缺乏可靠的相应参考基因组的新测序基因组中注释TE的机会。
挑战与机会
2010年对上一代注释工具进行了审核,认为必须以互补的方式运行多个工具才能获得详尽的注释。例如,被分类为重复的人类基因组的比例根据使用的工具而变化,上图。虽然de novo工具有可能识别新的重复家族并报告缺少的TE实例,但它们也更容易出现误报出假阳性的结果。仅依赖原始reads的方法在人类基因组中返回较低的重复内容,但它们具有能够在缺乏组装基因组的物种中注释TE的优点。最近对TE授权工具的比较研究也突出了当前基因组注释中缺少了的某些TE的注释。导致注释不完全。
检测TE多样性的工具
一旦参考基因组可用于注释,可以通过对不同个体进行测序来检测TE多态性(即,与参考基因组相比的TE的插入和缺失)。与单核苷酸多态性(SNP)相似,这种多态性插入可能与不同的表型相关,有些已经与血友病和Rett综合征等疾病有关。。许多软件工具都有开发用于检测种系和体细胞TE插入(具体可以参考原文中表三)。
用short reads 检测TE的多态性
大多数TE检测工作流程使用NGS数据作为输入。 NGS读数的长度通常为100-250个核苷酸,可以是单或双末端reads。使用这些短reads寻找新的插入是一个挑战,通常由三种策略中的一种或组合来解决:使用分裂reads(SR)信息,依赖于不一致的read对(DRP)或识别特定于TE的motif。这几个方法可以通过下图去理解。
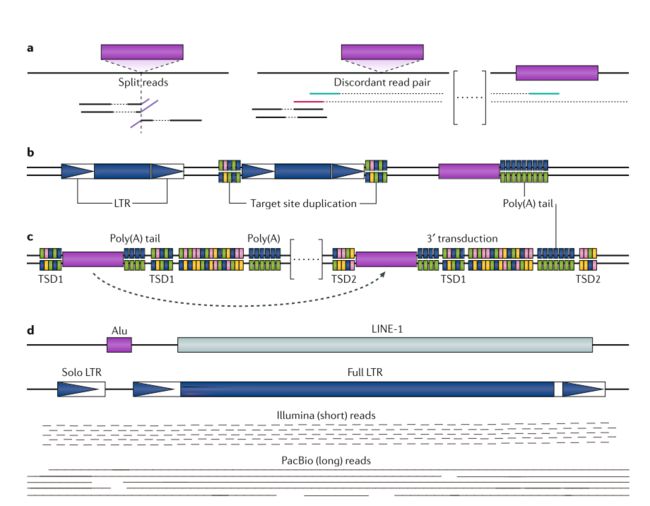
SR方法检测包含与插入位点重叠的断点的reads,其中read的一个段与参考比对,其余段包含TE序列的开序列始或终止序列。
DRP策略查看配对末端reads和标记,其中两个末端彼此远离排列,处于相反方向或者只有一个read在参考基因组上比对。DRP和SR分析可以作为初步的检测步骤或预先选择的TE比对的reads。总的来说SR方法更加精确,DRP方法更加灵敏。最好的方法是将两者结合,但是目前能够结合两种策略的工具不多。
第三种策略,检测工具是基于一定的TE motif在测序read中寻找已知的TE序列,不同于SR和DRP方法,它们是基于比对的方式。
不同的motif可以与TE相关联,例如LTR,TIRs。基于这几种不同的策略,工具可以检测个体和参考基因组之间的TE的多样性。短reads。不止可以用于生殖系可遗传的TE探测,也能用于体细胞的TE探测。目前一系列工具已经被应用于癌症细胞TE的探测。
基于捕获的short reads方法
通过靶向特定TE序列的DNA捕获方式也可以促进TE多态性的鉴定。
这。这个方法基,能够降低测序成本并丰富DNA的reads,这是TE检测的最有用信息方法可以产生足够的TE片段和结合的reads,以检测仅存在于少数细胞中的插入,并已用于鉴定癌症中的体细胞TE插入
长reads策略
最后,避免通过short reads和比对来探测TE插入所产生的误差的替代方案是利用最新发展的长reads测序技术。
实际上,长reads可以跨越整个TE插入以促进其检测,并且有可能检测甚至更复杂的结构变异事件,例如嵌套和串联插入,重复和反转。PacBio测序中的LoRTE就是其中一个例子,利用这个技术在人类中发现了2664个TE的插入事件,然后利用short reads只发现了893个插入事件。
这个工具是基于长
reads TE共有序列,针对参考基因组搜索那些reads的侧翼序列,并将来自相同方向和位置的侧翼对的内部序列与共有序列进行比较。一般对比short reads,long reads可以提供更好更准确的下游功能分析还有相关生物学的验证。
目前的挑战
尽管short reads 检测TE变得越来越popular了,但是其结果还没有展现很明显的一致性(工具间差异性比较大),所以要考虑将不同的工具相结合的方法。short reads取捕获方法,结合湿实验室TE富集和定制生物信息学分析,如RC-seq和TE-NGS,提供更高的灵敏度,但需要对目标区域进行排序。最后long readsTE检测工具有可能解决更多的插入和复杂的重排;但是,相关数据集及实验仍然很少。