通过前期的基因定量,我们通过RSEM或者Kallisto这些工具得到了每个基因或者转录本的表达量矩阵。输出的矩阵包含了基因以及在各个样本中的表达量信息,这些信息将为我们后期判定基因表达量变化趋势以及变化的显著性提供原始参考。
Trinity工具包同样也提供了一系列的脚本用以处理这些基因表达量变化差异显著性分析,在Trinity中主要使用的是EdgeR包进行处理。
在此之前我们需要注意一点:对于基因表达量分析由于是要对比不同样本,因此需要每个基因在不同样本中的表达量均有记录(哪怕是无表达量的0也可)。因此,Trinity建议在计算基因表达量时采用的参考转录本库应该是一个库,即先通过所有样本原始数据进行拼接而得到统一的转录本库,进而依照该转录本库进行回帖计算表达量。
在这个工作之前需要两个数据:
1. 基因表达的counts.matrix 文件
2. 生物学重复的表文件(由于后期比较的时候采用的配对显著性验证,因此会将每个样本之间进行比较,而存在生物学重复的类型需要通过该文件指明生物学重复情况)
此外,由于Trinity调用了多个R包,因此需要在进行运算前安装这些R包
% R
> source("http://bioconductor.org/biocLite.R") #由于包都是Bioconductor上的,因此需要安装这个东西
> biocLite('edgeR')
> biocLite('limma')
> biocLite('DESeq2')
> biocLite('ctc')
> biocLite('Biobase')
> install.packages('gplots')
> install.packages('ape')
接下来获取脚本帮助信息
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout$ $TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl -h
#################################################################################################
#
# Required:
#
# --matrix|m matrix of raw read counts (not normalized!)
#
# --method edgeR|DESeq2|voom|ROTS
# note: you should have biological replicates.
# edgeR will support having no bio replicates with
# a fixed dispersion setting.
#
# Optional:
#
# --samples_file|s tab-delimited text file indicating biological replicate relationships.
# ex.
# cond_A cond_A_rep1
# cond_A cond_A_rep2
# cond_B cond_B_rep1
# cond_B cond_B_rep2
#
#
# General options:
#
# --min_reps_min_cpm default: 2,1 (format: 'min_reps,min_cpm')
# At least min count of replicates must have cpm values > min cpm value.
# (ie. filtMatrix = matrix[rowSums(cpm(matrix)> min_cpm) >= min_reps, ] adapted from edgeR manual)
# Note, ** if no --samples_file, default for min_reps is set = 1 **
#
# --output|o name of directory to place outputs (default: $method.$pid.dir)
#
# --reference_sample name of a sample to which all other samples should be compared.
# (default is doing all pairwise-comparisons among samples)
#
# --contrasts file (tab-delimited) containing the pairs of sample comparisons to perform.
# ex.
# cond_A cond_B
# cond_Y cond_Z
#
#
###############################################################################################
#
# ## EdgeR-related parameters
# ## (no biological replicates)
#
# --dispersion edgeR dispersion value (Read edgeR manual to guide your value choice)
# http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
# ## ROTS parameters
# --ROTS_B : number of bootstraps and permutation resampling (default: 500)
# --ROTS_K : largest top genes size (default: 5000)
#
#
###############################################################################################
#
# Documentation and manuals for various DE methods. Please read for more advanced and more
# fine-tuned DE analysis than provided by this helper script.
#
# edgeR: http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
# DESeq2: http://bioconductor.org/packages/release/bioc/html/DESeq2.html
# voom/limma: http://bioconductor.org/packages/release/bioc/html/limma.html
# ROTS: http://www.btk.fi/research/research-groups/elo/software/rots/
#
###############################################################################################
可以看到Trinity提供的这个脚本是可以支持edgeR、DESeq2、voom/limma以及ROTS的,也就是说后期需要指定使用哪个软件进行分析。
EdgeR软件可以支持没有生物学重复的实验数据。通过指定dispersion参数进行设定,该参数很重要需要参考edgeR的使用说明进行设定。
我们的数据是有生物学重复的因此采用以下脚本进行:
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout$ cat samples.txt
B25 B251
B25 B252
R25 R251
R25 R252
W25 W251
W25 W252
#查看样品信息三个处理两个生物学重复
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ $TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl \#调用脚本
--matrix RSEM.isoform.counts.matrix \#采用的表达矩阵
--method edgeR \#采用的比对方式
--samples_file samples.txt \#
--output DEEdgeRout/ #结果输出到一个单独的文件夹
运行完了过后我们就去看看结果
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout$ cd DEEdgeRout/yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ l
RSEM.isoform.counts.matrix.B25_vs_R25.B25.vs.R25.EdgeR.Rscript
RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.count_matrix
RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results
RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results.MA_n_Volcano.pdf
RSEM.isoform.counts.matrix.B25_vs_W25.B25.vs.W25.EdgeR.Rscript
RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.count_matrix
RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results
RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results.MA_n_Volcano.pdf
RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.count_matrix
RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results
RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results.MA_n_Volcano.pdf
RSEM.isoform.counts.matrix.R25_vs_W25.R25.vs.W25.EdgeR.Rscript
12个文件共4类 包括
- Rscript(包括分析的脚本,通过这个脚本的修改可以调整参数和输出的图片)
- cout_matrix (提取出来的两两比对的数据)
- DE_results (这个是比对结果)
- PDF (可视化的图片)
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ head RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results
sampleA sampleB logFC logCPM PValue FDR
TRINITY_DN4700_c2_g1_i7 B25 R25 10.6165324802447 7.00741992505597 1.40250971832104e-141 7.67551493545557e-137
TRINITY_DN494_c0_g1_i9 B25 R25 11.3576752267624 6.27009842572613 5.23345757616711e-124 1.43205716385449e-119
TRINITY_DN26881_c0_g1_i3 B25 R25 14.7687111138408 6.44776948986216 1.07843333864274e-123 1.96731404413004e-119
TRINITY_DN5992_c0_g1_i13 B25 R25 14.5089667939309 6.1883806816389 6.15524143204199e-119 8.42144744628405e-115
TRINITY_DN1935_c0_g1_i1 B25 R25 -14.0852144942388 5.76481253295512 2.09305509819126e-103 2.29093252717426e-99
TRINITY_DN7856_c0_g1_i1 B25 R25 9.07630178998634 5.87159238635241 2.59525133714949e-102 2.36717199880301e-98
TRINITY_DN9161_c0_g1_i4 B25 R25 14.0055230986592 5.6858177658871 7.69816890064996e-1016.01853842036958e-97
TRINITY_DN41_c0_g1_i1 B25 R25 10.2304801828419 6.36856943794737 7.12383309140778e-99 4.87332516991842e-95
TRINITY_DN6913_c0_g1_i10 B25 R25 -13.8141572932327 5.49418010251322 1.17661182106672e-89 7.15471501461318e-86
关于FDR的含义与解释参考 https://www.jianshu.com/p/26511d3360c8

MAplot.png
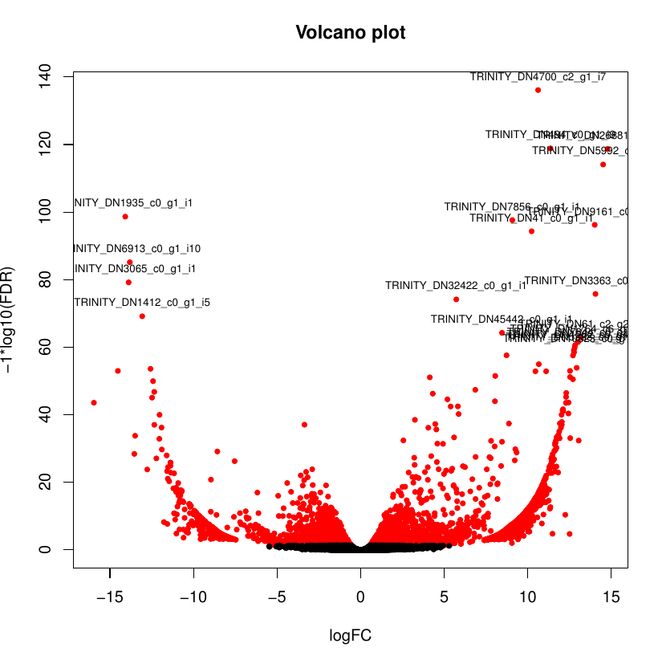
火山图.png
关于这个图的解释,参考 https://www.jianshu.com/p/26511d3360c8
接下来对差异数据进行注释和处理
首先绘制差异基因的热图以及样本分布
获取处理脚本的帮助文件
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ $TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl --help
####################################################################################
#
# Required:
#
# --matrix|m TMM.EXPR.matrix
#
# Optional:
#
# -P p-value cutoff for FDR (default: 0.001)
#
# -C min abs(log2(a/b)) fold change (default: 2 (meaning 2^(2) or 4-fold).
#
# --output prefix for output file (default: "diffExpr.P${Pvalue}_C${C})
#
#
#
#
# Misc:
#
# --samples|s sample-to-replicate mappings (provided to run_DE_analysis.pl)
#
# --max_DE_genes_per_comparison extract only up to the top number of DE features within each pairwise comparison.
# This is useful when you have massive numbers of DE features but still want to make
# useful heatmaps and other plots with more manageable numbers of data points.
#
# --order_columns_by_samples_file instead of clustering samples or replicates hierarchically based on gene expression patterns,
# order columns according to order in the --samples file.
#
# --max_genes_clust default: 10000 (if more than that, heatmaps are not generated, since too time consuming)
#
# --examine_GO_enrichment run GO enrichment analysis
# --GO_annots GO annotations file
# --gene_lengths lengths of genes file
#
# --include_GOplot optional: will generate inputs to GOplot and attempt to make a preliminary pdf plot/report for it.
#
##############################################################
运行这个脚本进行计算后的得到结果
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ l -alt
total 76916
-rw-rw-r-- 1 yeyuntian yeyuntian 52233286 1月 29 17:21 diffExpr.P0.001_C2.matrix.RData
drwxrwxr-x 2 yeyuntian yeyuntian 4096 1月 29 17:21 ./
-rw-rw-r-- 1 yeyuntian yeyuntian 206154 1月 29 17:21 diffExpr.P0.001_C2.matrix.log2.centered.genes_vs_samples_heatmap.pdf
-rw-rw-r-- 1 yeyuntian yeyuntian 6462 1月 29 17:21 diffExpr.P0.001_C2.matrix.log2.centered.sample_cor_matrix.pdf
-rw-rw-r-- 1 yeyuntian yeyuntian 626 1月 29 17:21 diffExpr.P0.001_C2.matrix.log2.centered.sample_cor.dat
-rw-rw-r-- 1 yeyuntian yeyuntian 490031 1月 29 17:21 diffExpr.P0.001_C2.matrix.log2.centered.dat
-rw-rw-r-- 1 yeyuntian yeyuntian 4611 1月 29 17:21 diffExpr.P0.001_C2.matrix.R
-rw-rw-r-- 1 yeyuntian yeyuntian 224690 1月 29 17:21 diffExpr.P0.001_C2.matrix
-rw-rw-r-- 1 yeyuntian yeyuntian 59 1月 29 17:21 DE_feature_counts.P0.001_C2.matrix
-rw-rw-r-- 1 yeyuntian yeyuntian 58609 1月 29 17:21 RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results.P0.001_C2.DE.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 23967 1月 29 17:21 RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results.P0.001_C2.R25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 34712 1月 29 17:21 RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results.P0.001_C2.W25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 239536 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results.P0.001_C2.B25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 400145 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results.P0.001_C2.DE.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 160679 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results.P0.001_C2.W25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 36 1月 29 17:21 RSEM.isoform.counts.matrix.R25_vs_W25.edgeR.DE_results.samples
-rw-rw-r-- 1 yeyuntian yeyuntian 36 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_W25.edgeR.DE_results.samples
-rw-rw-r-- 1 yeyuntian yeyuntian 219363 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results.P0.001_C2.B25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 317428 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results.P0.001_C2.DE.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 98135 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results.P0.001_C2.R25-UP.subset
-rw-rw-r-- 1 yeyuntian yeyuntian 36 1月 29 17:21 RSEM.isoform.counts.matrix.B25_vs_R25.edgeR.DE_results.samples
在这些结果包括
- 两两比较的样本信息(.samples)
- 样本比较后的基因表达情况上调还是下调的文件以及整合在一起的基因(.subset)
- 差异基因在不同样本中分布数量(DE_feature_counts.P0.001_C2.matrix)
- 差异基因表达量矩阵(diffExpr.P0.001_C2.matrix)
- 差异基因对数转换后的表达矩阵 (diffExpr.P0.001_C2.matrix.log2.centered.dat)
-
出的两个图(PDF)
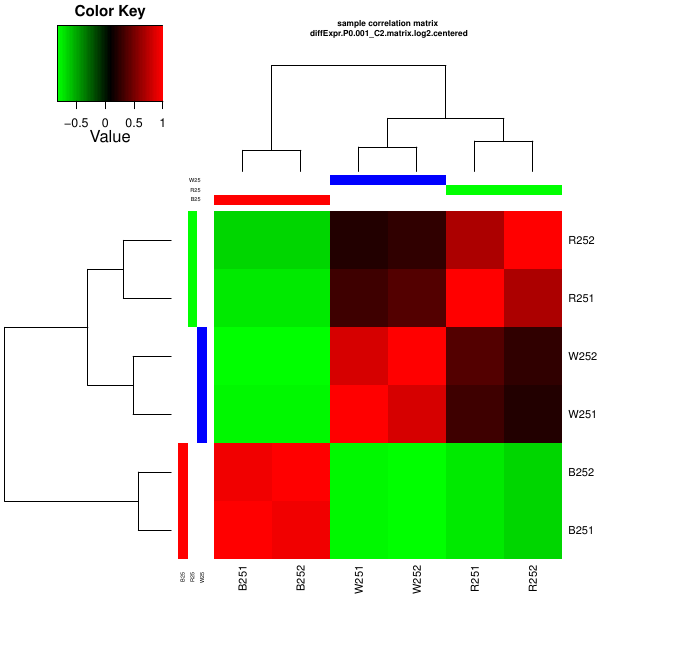 image.png
image.png
 image.png
image.png - 包括一个R脚本和一个Rdata(这两个文件可以通过修改和调用实现对后期出图的控制)
最后要提示一点就是注意对于R脚本修改后可以通过shell在所在文件夹进行直接运行后得到对应的图片
yeyuntian@yeyuntian-RESCUER-R720-15IKBN:~/Biodata/trinitytest/downstr/RSEMout/RSEMout/DEEdgeRout$ Rscript diffExpr.P0.001_C2.matrix.R