在上一篇文章PH525x series - Bayesian Statistics中是将层次模型应用到了棒球运动当中,这篇文章作者讲的是在高通量数据的分析中如何应用层次模型。
既然是层次模型,那我们首先要做的就是构建两个,第一个描述的是样本或单元之间的变异,第二个则描述的是某种特征之间的变异。这次作者使用的例子是limma包中的一组基因表达数据(2组,每组3个样本,其中共有16个差异显著基因),这个包提供了一种修饰,比原本的更具有统计效力。
与棒球运动的例子不同的是,这次我们为方差建模。我们先来看使用原本的检验做差异显著检验的结果是怎样的:
library(SpikeInSubset) ##Available from Bioconductor data(rma95)
library(genefilter)
fac <- factor(rep(1:2,each=3))
tt <- rowttests(exprs(rma95),fac)
smallp <- with(tt, p.value < .01)
#注意pData(rma95)获得的那些基因是真实存在差异的基因,共16个
spike <- rownames(rma95) %in% colnames(pData(rma95))
cols <- ifelse(spike,"dodgerblue",ifelse(smallp,"red","black"))
with(tt, plot(-dm, -log10(p.value), cex=.8, pch=16,
xlim=c(-1,1), ylim=c(0,4.5),
xlab="difference in means",
col=cols))
abline(h=2,v=c(-.2,.2), lty=2)

上述结果有两点问题,这第一便是使用FDR对值进行检验之后,仅剩1个基差异显著的基因;
sum(p.adjust(tt$p.value,method="BH")[spike] < 0.05)
## [1] 1
当然导致这一结果的原因肯定与样本量过少有关。第二个问题就是按照的值对基因进行排序后,无论是取排名前多少的一组基因,这当中都包含了很多的假阳性,比如在排名前10的基因中有6个是假阳性:
table(top10 =rank(tt$p.value <=10, spike)) #t-stat and p-val rank is the same
## spike
## top10 FALSE TRUE
## FALSE 12604 12
## TRUE 6 4
从上面的火山图中我们可以看到,小p值但又是假阳性的那些基因其实就是那些效应力很小(也就是标准误会很小)的基因(图中那些红点其横坐标集中在了)。我们可以通过下图确认这点:
tt$s <- apply(exprs(rma95), 1, function(row) sqrt(.5 * (var(row[1:3]) + var(row[4:6]) ) ) )
with(tt, plot(s, -log10(p.value), cex=.8, pch=16,
log="x",xlab="estimate of standard deviation",col=cols))

这种情况下,使用层次模型会更有效。如果我们能够为这些基因的方差构建分布模型,就可以根据这种分布修饰那些过小的p值,从而达到提高统计效力的目的。
根据PH525x series - Statistical Models(二)中介绍的分布可知,我们可以使用如下模型为方差建模:
像我们这种利用经验(empirical)数据构建先验分布(Bayesian approach)的方法就是经验贝叶斯方法。现在我们有了每个基因的观察值,也有了先验分布模型(即F分布),按照如下公式公式就可以去计算方差的后验分布平均值了:
在这里,是观察值的方差(observed variance,就是实际的方差),是全局方差(global variance,其实就是后验分布的方差,也就是由果求因得到的真实的方差)。在得到后验分布的均值和标准差之后,我们就可以使用这两个数值去进行moderated t-tests了,这一过程说起来复杂,代码其实就几行:
library(limma)
##先为每个基因构建一个线性模型
fit <- lmFit(rma95, model.matrix(~ fac))
##进行经验贝叶斯检验
ebfit <- eBayes(fit)
##提取差值与p值
limmares <- data.frame(dm=coef(fit)[,"fac2"], p.value=ebfit$p.value[,"fac2"])
##绘图
with(limmares, plot(dm, -log10(p.value),cex=.8, pch=16,
col=cols,xlab="difference in means",
xlim=c(-1,1), ylim=c(0,5)))
abline(h=2,v=c(-.2,.2), lty=2)
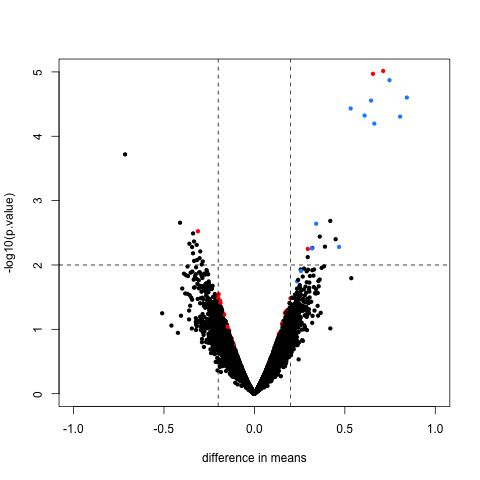
这样一来,在p值升序排列的前10个基因中假阳性率就降到了2:
table( top10=rank(limmares$p.value)<= 10, spike)
## spike
## top50 FALSE TRUE
## FALSE 12608 8
## TRUE 2 8
可见这种moderated t-tests的统计效力要比原始的** t-tests**要好。还有一个重要的点要知道:原先那些标准差接近0的样本在通过层次模型矫正后就不再接近0了,而是朝着的方向收缩。
函数讲解:
limFit(object): Fit linear model for each gene given a series of arraysebayes(fit): Given a microarray linear model fit, compute moderated t-statistics, moderated F-statistic, and log-odds of differential expression by empirical Bayes moderation of the standard errors towards a common value.coef(object): ‘coef’ is a generic function which extracts model coefficients from objects returned by modeling functions.
原文请戳