代码分析 | 单细胞转录组数据整合详解
两种整合方法详解
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
单细胞转录组数据整合的方法多种多样,当然软件也是层出不穷,有的感觉矫正效应过强,导致不是一种细胞类型的细胞也会整合在一起,让人难以评判。前段时间有同学问我她有不同人相同肿瘤的样本,问我应该使用Merge还是使用CCA(单细胞分析Seurat使用相关的10个问题答疑精选!),我只能说实话我是不知道的,如果是我我都会试一试。由于肿瘤细胞的异质性过强,并且具有极大的样本差异性,如果使用CCA等进行整合,不知道会不会影响分析结果。并且在文章中我看的往往肿瘤样本会因为异质性导致无法重合,但其微环境如T、B等细胞还是可以有效的整合在一起,所以,保险起见,try a lot,select one!
本次接着上两节进行的芬兰CSC-IT科学中心主讲的生物信息课程(https://www.csc.fi/web/training/-/scrnaseq)视频,官网上还提供了练习素材以及详细代码,今天就来练习一下单细胞数据整合的过程。
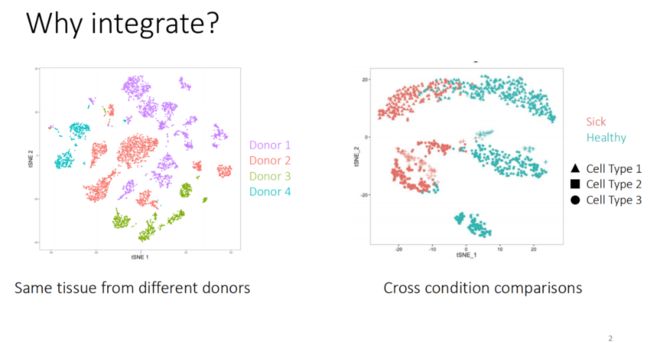
在本教程中将探讨不同的整合多个单细胞RNA-seq数据集方法。我们将探索两种不同方法的校正整个数据集批次效应的效果并定量评估整合数据的质量。
数据集
在本教程中,我们将使用来自四种技术的3种不同的人类胰岛细胞数据集:CelSeq(GSE81076)、CelSeq2(GSE85241)、Fluidigm C1(GSE86469)和SMART-Seq2(E-MTAB-5061)。
原始数据矩阵和metadata下载链接:
https://www.dropbox.com/s/1zxbn92y5du9pu0/pancreas_v3_files.tar.gz?dl=1
加载所需R包
suppressMessages(require(Seurat))
suppressMessages(require(ggplot2))
suppressMessages(require(cowplot))
suppressMessages(require(scater))
suppressMessages(require(scran))
suppressMessages(require(BiocParallel))suppressMessages(require(BiocNeighbors))Seurat (anchors and CCA)
我们将使用在文章Comprehensive Integration of Single Cell Data[1]中所提到的数据整合方法。
数据处理
加载表达式矩阵和metadata。metadata文件包含四个数据集中每个细胞所用技术平台和细胞类型注释。
pancreas.data <- readRDS(file = "session-integration_files/pancreas_expression_matrix.rds")metadata <- readRDS(file = "session-integration_files/pancreas_metadata.rds")创建具有所有数据集的Seurat对象。
pancreas <- CreateSeuratObject(pancreas.data, meta.data = metadata)在应用任何批次校正之前先看一下数据集。我们先执行标准预处理(log-normalization)并基于方差稳定化转换(“vst”)识别变量特征,接下来对集成数据进行归一化、运行PCA并使用UMAP可视化结果。集成数据集是按细胞类型而不是测序平台进行聚类。
# 标准化并查找可变基因
pancreas <- NormalizeData(pancreas, verbose = FALSE)
pancreas <- FindVariableFeatures(pancreas, selection.method = "vst", nfeatures = 2000, verbose = FALSE)
# 运行标准流程并进行可视化
pancreas <- ScaleData(pancreas, verbose = FALSE)
pancreas <- RunPCA(pancreas, npcs = 30, verbose = FALSE)
pancreas <- RunUMAP(pancreas, reduction = "pca", dims = 1:30)
p1 <- DimPlot(pancreas, reduction = "umap", group.by = "tech")
p2 <- DimPlot(pancreas, reduction = "umap", group.by = "celltype", label = TRUE, repel = TRUE) +
NoLegend()plot_grid(p1, p2)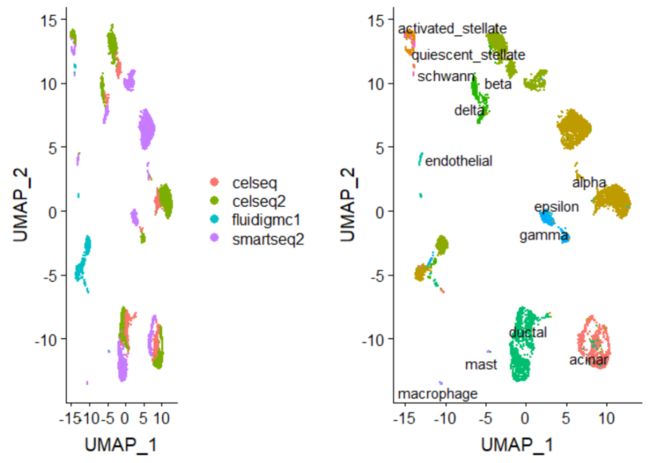
将合并的对象分成一个列表,每个数据集都作为一个元素。通过执行标准预处理(log-normalization)并基于方差稳定化转换(“vst”)分别为每个数据集查找变化的基因。
pancreas.list <- SplitObject(pancreas, split.by = "tech")
for (i in 1:length(pancreas.list)) {
pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)
pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst", nfeatures = 2000,
verbose = FALSE)}4个胰岛细胞数据集的整合
使用FindIntegrationAnchors函数来识别锚点(anchors),该函数的输入数据是Seurat对象的列表。
reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2", "fluidigmc1")]pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, dims = 1:30)## Computing 2000 integration features
## Scaling features for provided objects
## Finding all pairwise anchors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 3499 anchors
## Filtering anchors
## Retained 2821 anchors
## Extracting within-dataset neighbors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 3515 anchors
## Filtering anchors
## Retained 2701 anchors
## Extracting within-dataset neighbors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 6173 anchors
## Filtering anchors
## Retained 4634 anchors
## Extracting within-dataset neighbors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 2176 anchors
## Filtering anchors
## Retained 1841 anchors
## Extracting within-dataset neighbors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 2774 anchors
## Filtering anchors
## Retained 2478 anchors
## Extracting within-dataset neighbors
## Running CCA
## Merging objects
## Finding neighborhoods
## Finding anchors
## Found 2723 anchors
## Filtering anchors
## Retained 2410 anchors
## Extracting within-dataset neighbors然后将这些锚(anchors)传递给IntegrateData函数,该函数返回Seurat对象。
pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)## Merging dataset 4 into 2
## Extracting anchors for merged samples
## Finding integration vectors
## Finding integration vector weights
## Integrating data
## Merging dataset 1 into 2 4
## Extracting anchors for merged samples
## Finding integration vectors
## Finding integration vector weights
## Integrating data
## Merging dataset 3 into 2 4 1
## Extracting anchors for merged samples
## Finding integration vectors
## Finding integration vector weights
## Integrating data运行IntegrateData之后,Seurat对象将包含一个具有整合(或“批次校正”)表达矩阵的新Assay。请注意,原始值(未校正的值)仍存储在“RNA”分析的对象中,因此可以来回切换。
然后可以使用这个新的整合矩阵进行下游分析和可视化,我们在这里做了整合数据标准化、运行PCA并使用UMAP可视化结果。
# switch to integrated assay. The variable features of this assay are automatically set during
# IntegrateData
DefaultAssay(pancreas.integrated) <- "integrated"
# 运行标准流程并进行可视化
pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)
pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)
pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)
p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")
p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE, repel = TRUE) +
NoLegend()plot_grid(p1, p2)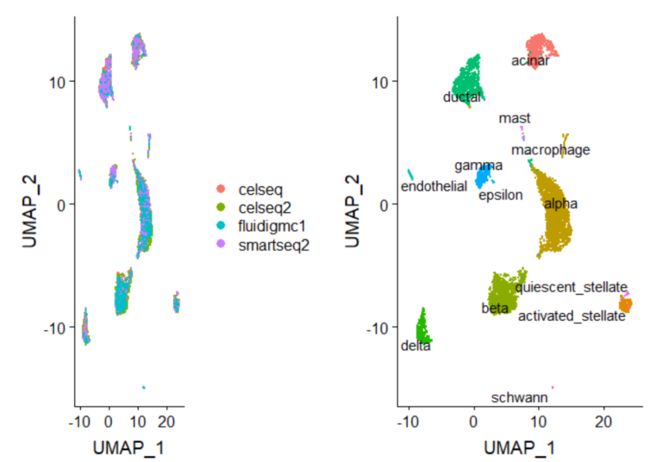
Mutual Nearest Neighbor (MNN)
整合单细胞RNA-seq数据的另一种方法是使用相互最近邻(MNN)批次校正方法,可参考Haghverdi et al[2]。
首先可以直接从计数矩阵创建SingleCellExperiment(SCE)对象,也可以直接从Seurat转换为SCE。
celseq.data <- as.SingleCellExperiment(pancreas.list$celseq)
celseq2.data <- as.SingleCellExperiment(pancreas.list$celseq2)
fluidigmc1.data <- as.SingleCellExperiment(pancreas.list$fluidigmc1)smartseq2.data <- as.SingleCellExperiment(pancreas.list$smartseq2)数据处理
查找共同基因并将每个数据集简化为那些共同基因:
keep_genes <- Reduce(intersect, list(rownames(celseq.data),rownames(celseq2.data),
rownames(fluidigmc1.data),rownames(smartseq2.data)))
celseq.data <- celseq.data[match(keep_genes, rownames(celseq.data)), ]
celseq2.data <- celseq2.data[match(keep_genes, rownames(celseq2.data)), ]
fluidigmc1.data <- fluidigmc1.data[match(keep_genes, rownames(fluidigmc1.data)), ]smartseq2.data <- smartseq2.data[match(keep_genes, rownames(smartseq2.data)), ]通过查找具有异常低的总计数或特征(基因)总数的异常值,使用calculateQCMetrics()计算QC来确定低质量细胞(对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择)。
## celseq.data
celseq.data <- calculateQCMetrics(celseq.data)
low_lib_celseq.data <- isOutlier(celseq.data$log10_total_counts, type="lower", nmad=3)
low_genes_celseq.data <- isOutlier(celseq.data$log10_total_features_by_counts, type="lower", nmad=3)
celseq.data <- celseq.data[, !(low_lib_celseq.data | low_genes_celseq.data)]
## celseq2.data
celseq2.data <- calculateQCMetrics(celseq2.data)
low_lib_celseq2.data <- isOutlier(celseq2.data$log10_total_counts, type="lower", nmad=3)
low_genes_celseq2.data <- isOutlier(celseq2.data$log10_total_features_by_counts, type="lower", nmad=3)
celseq2.data <- celseq2.data[, !(low_lib_celseq2.data | low_genes_celseq2.data)]
## fluidigmc1.data
fluidigmc1.data <- calculateQCMetrics(fluidigmc1.data)
low_lib_fluidigmc1.data <- isOutlier(fluidigmc1.data$log10_total_counts, type="lower", nmad=3)
low_genes_fluidigmc1.data <- isOutlier(fluidigmc1.data$log10_total_features_by_counts, type="lower", nmad=3)
fluidigmc1.data <- fluidigmc1.data[, !(low_lib_fluidigmc1.data | low_genes_fluidigmc1.data)]
## smartseq2.data
smartseq2.data <- calculateQCMetrics(smartseq2.data)
low_lib_smartseq2.data <- isOutlier(smartseq2.data$log10_total_counts, type="lower", nmad=3)
low_genes_smartseq2.data <- isOutlier(smartseq2.data$log10_total_features_by_counts, type="lower", nmad=3)smartseq2.data <- smartseq2.data[, !(low_lib_smartseq2.data | low_genes_smartseq2.data)]使用scran包的computeSumFactors()和normalize()函数计算大小(size)并对数据进行标准化:
# 计算尺寸因子(sizefactors)
celseq.data <- computeSumFactors(celseq.data)
celseq2.data <- computeSumFactors(celseq2.data)
fluidigmc1.data <- computeSumFactors(fluidigmc1.data)
smartseq2.data <- computeSumFactors(smartseq2.data)
# 标准化
celseq.data <- normalize(celseq.data)
celseq2.data <- normalize(celseq2.data)
fluidigmc1.data <- normalize(fluidigmc1.data)smartseq2.data <- normalize(smartseq2.data)特征选择:我们使用TrendVar()和decomposeVar()函数来计算每个基因的变异(variance),并将其分为技术平台和生物两个部分的差异。
## celseq.data
fit_celseq.data <- trendVar(celseq.data, use.spikes=FALSE)
dec_celseq.data <- decomposeVar(celseq.data, fit_celseq.data)
dec_celseq.data$Symbol_TENx <- rowData(celseq.data)$Symbol_TENx
dec_celseq.data <- dec_celseq.data[order(dec_celseq.data$bio, decreasing = TRUE), ]
## celseq2.data
fit_celseq2.data <- trendVar(celseq2.data, use.spikes=FALSE)
dec_celseq2.data <- decomposeVar(celseq2.data, fit_celseq2.data)
dec_celseq2.data$Symbol_TENx <- rowData(celseq2.data)$Symbol_TENx
dec_celseq2.data <- dec_celseq2.data[order(dec_celseq2.data$bio, decreasing = TRUE), ]
## fluidigmc1.data
fit_fluidigmc1.data <- trendVar(fluidigmc1.data, use.spikes=FALSE)
dec_fluidigmc1.data <- decomposeVar(fluidigmc1.data, fit_fluidigmc1.data)
dec_fluidigmc1.data$Symbol_TENx <- rowData(fluidigmc1.data)$Symbol_TENx
dec_fluidigmc1.data <- dec_fluidigmc1.data[order(dec_fluidigmc1.data$bio, decreasing = TRUE), ]
## smartseq2.data
fit_smartseq2.data <- trendVar(smartseq2.data, use.spikes=FALSE)
dec_smartseq2.data <- decomposeVar(smartseq2.data, fit_smartseq2.data)
dec_smartseq2.data$Symbol_TENx <- rowData(smartseq2.data)$Symbol_TENx
dec_smartseq2.data <- dec_smartseq2.data[order(dec_smartseq2.data$bio, decreasing = TRUE), ]
#选择在所有数据集中共有的且信息最丰富的基因:
universe <- Reduce(intersect, list(rownames(dec_celseq.data),rownames(dec_celseq2.data),
rownames(dec_fluidigmc1.data),rownames(dec_smartseq2.data)))
mean.bio <- (dec_celseq.data[universe,"bio"] + dec_celseq2.data[universe,"bio"] +
dec_fluidigmc1.data[universe,"bio"] + dec_smartseq2.data[universe,"bio"])/4hvg_genes <- universe[mean.bio > 0]将数据集合并到SingleCellExperiment中:
# 总原始counts的整合
counts_pancreas <- cbind(counts(celseq.data), counts(celseq2.data),
counts(fluidigmc1.data), counts(smartseq2.data))
# 总的标准化后的counts整合 (with multibatch normalization)
logcounts_pancreas <- cbind(logcounts(celseq.data), logcounts(celseq2.data),
logcounts(fluidigmc1.data), logcounts(smartseq2.data))
# 构建整合数据的sce对象
sce <- SingleCellExperiment(
assays = list(counts = counts_pancreas, logcounts = logcounts_pancreas),
rowData = rowData(celseq.data), # same as rowData(pbmc4k)
colData = rbind(colData(celseq.data), colData(celseq2.data),
colData(fluidigmc1.data), colData(smartseq2.data))
)
# 将前面的hvg_genes存储到sce对象的metadata slot中:metadata(sce)$hvg_genes <- hvg_genes用MNN处理批次效应之前先看一下这些datasets:
sce <- runPCA(sce,
ncomponents = 20,
feature_set = hvg_genes,
method = "irlba")
names(reducedDims(sce)) <- "PCA_naive"
p1 <- plotReducedDim(sce, use_dimred = "PCA_naive", colour_by = "tech") +
ggtitle("PCA Without batch correction")
p2 <- plotReducedDim(sce, use_dimred = "PCA_naive", colour_by = "celltype") +
ggtitle("PCA Without batch correction")plot_grid(p1, p2)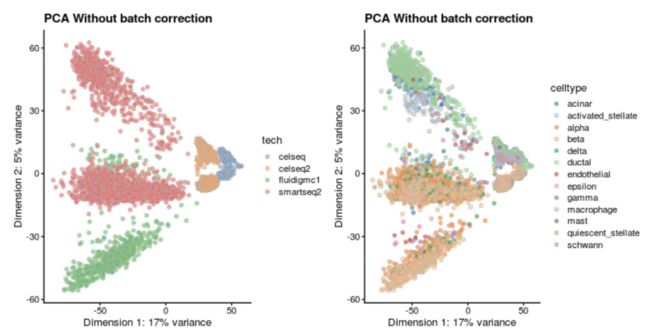
使用MNN进行数据整合
scran软件包中的MNN方法利用一种新方法来调整批次效应-fastMNN()。
fastMNN()函数返回的是降维数据表示形式,该表示形式的使用与其他较低维度的表示形式(例如PCA)类似。
跑fastMNN()之前,我们需要先rescale每一个批次,来调整不同批次之间的测序深度。用scran包里的multiBatchNorm()函数对size factor进行调整后,重新计算log标准化的表达值以适应不同SingleCellExperiment对象的系统差异。之前的size factors仅能移除单个批次里细胞之间的bias。现在我们通过消除批次之间技术差异来提高校正的质量。
rescaled <- multiBatchNorm(celseq.data, celseq2.data, fluidigmc1.data, smartseq2.data)
celseq.data_rescaled <- rescaled[[1]]
celseq2.data_rescaled <- rescaled[[2]]
fluidigmc1.data_rescaled <- rescaled[[3]]smartseq2.data_rescaled <- rescaled[[4]]
跑fastMNN,把降维的MNN representation存在sce对象的reducedDimsslot里:
mnn_out <- fastMNN(celseq.data_rescaled,
celseq2.data_rescaled,
fluidigmc1.data_rescaled,
smartseq2.data_rescaled,
subset.row = metadata(sce)$hvg_genes,
k = 20, d = 50, approximate = TRUE,
# BPPARAM = BiocParallel::MulticoreParam(8),
BNPARAM = BiocNeighbors::AnnoyParam())
reducedDim(sce, "MNN") <- mnn_out$correct注意:fastMNN()不会生成批次校正的表达矩阵。因此,fastMNN()的结果应仅被视为降维表示,适合直接绘图,如TSNE/ UMAP、聚类和依赖于此类结果的轨迹分析(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集)。
p1 <- plotReducedDim(sce, use_dimred = "MNN", colour_by = "tech") + ggtitle("MNN Ouput Reduced Dimensions")
p2 <- plotReducedDim(sce, use_dimred = "MNN", colour_by = "celltype") + ggtitle("MNN Ouput Reduced Dimensions")plot_grid(p1, p2)
Session info
sessionInfo()## R version 3.5.3 (2019-03-11)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 10 x64 (build 17763)
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=English_United States.1252
## [2] LC_CTYPE=English_United States.1252
## [3] LC_MONETARY=English_United States.1252
## [4] LC_NUMERIC=C
## [5] LC_TIME=English_United States.1252
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets## [8] methods base[1]:https://www.biorxiv.org/content/10.1101/460147v1[2]:https://www.nature.com/articles/nbt.4091
撰文:Tiger校对:生信宝典
你可能还想看
“harmony”整合不同平台的单细胞数据之旅
Cell 深度 一套普遍适用于各类单细胞测序数据集的锚定整合方案
重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
如何使用Bioconductor进行单细胞分析?
如何火眼金睛鉴定那些单细胞转录组中的混杂因素
什么?你做的差异基因方法不合适?