上一步骤(第6篇:重复样本的处理——IDR)用IDR对重复样本peaks的一致性进行了评估,同时得到了merge后的一致性的peaks——
sample-idr,接下来就是对peaks的注释。这篇主要介绍用Y叔的R包ChIPseeker对peaks的位置(如peaks位置落在启动子、UTR、内含子等)以及peaks临近基因的注释。
ChIPseeker
ChIPseeker虽然最初是为了ChIP-seq注释而写的一个R包,但它不只局限于ChIP-seq,也可用于ATAC-Seq等其他富集peaks注释,也可用于lincRNA注释、DNA breakpoints的断点位置注释等所有genomic coordination的注释,另外提供了丰富的可视化功能。
ChIPseeker的强大功能请参考Y叔的ChIP-Seq系列文章,如:
CS3: peak注释
CS4:关于ChIPseq注释的几个问题
CS6: ChIPseeker的可视化方法(中秋节的视觉饕餮)

使用方法
使用ChIPseeker需要准备两个文件:一个就是要注释的peaks的文件,需满足BED格式。另一个就是注释参考文件,即需要一个包含注释信息的TxDb对象。Bioconductor提供了30个TxDb包,如果其中有研究的物种就可以直接下载安装此物种的TxDb信息。如果研究的物种没有已知的TxDb,可以使用GenomicFeatures包的函数(makeTxDbFromUCSC,makeTxDbFromBiomart)制作TxDb对象:
makeTxDbFromUCSC: 通过UCSC在线制作TxDb
makeTxDbFromBiomart: 通过ensembl在线制作TxDb
makeTxDbFromGRanges:通过GRanges对象制作TxDb
makeTxDbFromGFF:通过解析GFF文件制作TxDb
制作TxDb方法示例(CS4:关于ChIPseq注释的几个问题):
- 如用人的参考基因信息来做注释,从UCSC生成TxDb:
library(GenomicFeatures)
hg19.refseq.db <- makeTxDbFromUCSC(genome="hg19", table="refGene")
- 用GFF文件做裂殖酵母的注释
download.file("ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosaccharomyces_pombe.chr.gff3", "schizosaccharomyces_pombe.chr.gff3")require(GenomicFeatures)
spombe <- makeTxDbFromGFF("schizosaccharomyces_pombe.chr.gff3")
具体步骤如下:
第1步:下载安装ChIPseeker注释相关的包
从Bioconductor直接下载,或从github安装最新版本
source ("https://bioconductor.org/biocLite.R")
biocLite("ChIPseeker")
# 下载人的基因和lincRNA的TxDb对象
biocLite("org.Hs.eg.db")
biocLite("TxDb.Hsapiens.UCSC.hg19.knownGene")
biocLite("TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts")
biocLite("clusterProfiler")
#载入各种包
library("ChIPseeker")
library(clusterProfiler)
library("org.Hs.eg.db")
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
library("TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts")
lincRNA_txdb=TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts
第2步:读入peaks文件
函数readPeakFile读入peaks文件
nanog <- readPeakFile("./idr_out.bed/nanog_idr-bed")
pou5f1 <- readPeakFile("./idr_out.bed/pou5f1_idr-bed")
第3步:注释peaks
peaks的注释是用的annotatePeak函数,可以单独对每个peaks文件进行注释,也可以将多个peaks制作成一个list,进行比较分析和可视化。
# 制作多个样本比较的list
peaks <- list(Nanog=nanog,Pou5f1=pou5f1)
# promotor区间范围可以自己设定
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrixList <- lapply(peaks, getTagMatrix, windows=promoter)
#annotatePeak传入annoDb参数,可进行基因ID转换(Entrez,ENSEMBL,SYMBOL,GENENAME)
peakAnnoList <- lapply(peaks, annotatePeak, TxDb=txdb,tssRegion=c(-3000, 3000), verbose=FALSE,addFlankGeneInfo=TRUE, flankDistance=5000,annoDb="org.Hs.eg.db")
annotatePeak传入annoDb参数,即可进行基因ID转换,将Entrez ID转化为ENSEMBL,SYMBOL,GENENAME,peakAnnoList的结果如下:
seqnames start end width strand annotation geneChr geneStart geneEnd geneLength geneStrand geneId transcriptId distanceToTSS ENSEMBL SYMBOL GENENAME flank_txIds flank_geneIds flank_gene_distances
5 chr3 196625522 196625873 352 * Intron (uc003fwz.4/205564, intron 2 of 9) 3 196594727 196661584 66858 1 205564 uc011bty.2 30795 ENSG00000119231 SENP5 SUMO specific peptidase 5 uc003fwz.4;uc011bty.2 205564;205564 0;0
第4步:可视化
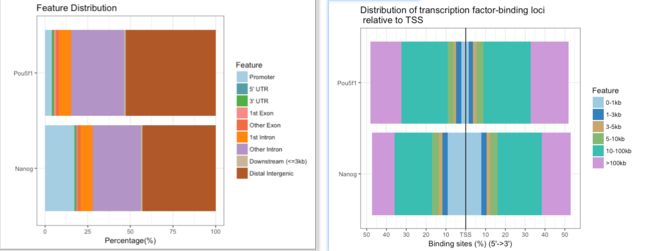
提供了多种可视化方法,如plotAnnoBar(),vennpie(),plotAnnoPie(),plotDistToTSS()等,下面展示了两个样本在基因组特征区域的分布以及转录因子在TSS区域的结合。
plotAnnoBar(peakAnnoList)
plotDistToTSS(peakAnnoList,title="Distribution of transcription factor-binding loci \n relative to TSS")
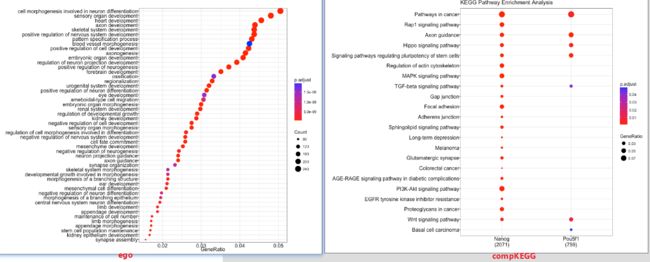
第5步:功能富集分析
提取peakAnnolist中的基因,结合clusterProfiler包对peaks内的邻近基因进行富集注释。
# Create a list with genes from each sample
gene = lapply(peakAnnoList, function(i) as.data.frame(i)$geneId)
# Run GO enrichment analysis
ego <- enrichGO(gene = entrez,
keytype = "ENTREZID",
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
readable = TRUE)
# Dotplot visualization
dotplot(ego, showCategory=50)
# Multiple samples KEGG analysis
compKEGG <- compareCluster(geneCluster = gene,
fun = "enrichKEGG",
organism = "human",
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
dotplot(compKEGG, showCategory = 20, title = "KEGG Pathway Enrichment Analysis")
第6步:保存文件
# Output peakAnnolist file
save(peakAnnoList,file="peakAnnolist.rda")
write.table(as.data.frame(peakAnnoList$Nanog),file="Nanog.PeakAnno",sep='\t',quote = F)
# Output results from GO analysis to a table
cluster_summary <- data.frame(ego)
write.csv(cluster_summary, "results/clusterProfiler_Nanog.csv")
参考资料:
ChIPseeker详细内容请参考:
- ChIPseeker文档
- Y叔的ChIP-Seq系列:
CS3: peak注释
CS4:关于ChIPseq注释的几个问题
CS6: ChIPseeker的可视化方法(中秋节的视觉饕餮) - 哈佛深度NGS数据分析课程
- 07-ChIPseeker for ChIP peak Annotation, Comparison, and Visualization