目录
- 写在前面
- 功能注释数据库介绍
- 方法一: 以KEGG的注释结果为主, 筛选出每个品种包含的特异通路及基因
- 方法二: 利用pfam的注释结果, 过滤掉每个品种具有相同结构域的基因, 对于剩下的基因, 利用其swissprot id 在DAVID数据库中查找他们的功能信息, 单个进行筛选
- 最后
写在前面
-
事情是这样的, 前段时间接到师兄布置的一个任务, 需要从9个品种特异区段的总共4000多个基因中找到跟特异性状相关的基因, 而且最好是单个找, 尽量不要漏掉每个信息, 然后我就拿到了这些基因的功能注释文件, 先上图吧
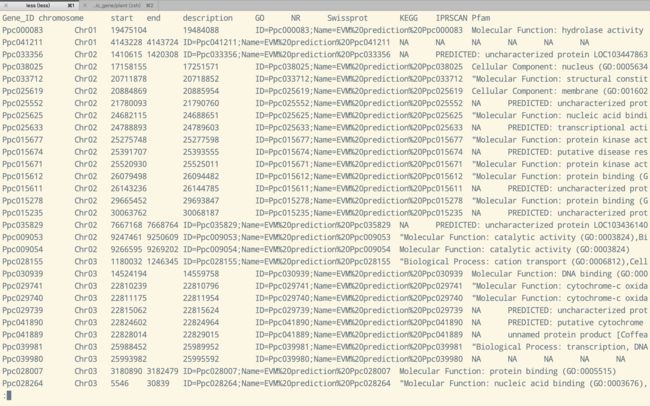 功能注释文件
功能注释文件 - 相信接触过基因组注释的同学对这个文件印象都很深刻, 这是折腾好多天整合多个数据库得到的功能注释结果, 这个结果中包含了GO, NCBI, Swissprot, KEGG, IPRSCAN和Pfam六个数据库
- 这篇笔记没有什么流程性可言, 只是我为了更快更好地完成任务想出来的小办法, 如果哪位大神觉得有问题, 希望能给我指出, 大家共同进步, 跪谢了~
功能注释数据库介绍
GO数据库
- Gene Ontology (GO)为了能够使对各种数据库中基因产物的功能描述相一致,发展了具有三级结构的标准语言(ontologies)。根据基因产物的相关分子功能,生物学途径,细胞学组成而给予定义,无物种相关性。目前,GO的定义法则已经在多个合作的数据库中使用,使得这些数据库在查询时具有很高的一致性
- 在基因表达分析中GO数据库如何发挥它的作用呢?很简单,将转录组数据引入GO注释,可以揭示某一特定组具有相似表达模式基因的功能,因为共表达的基因可能编码在同一个生物过程中出现的基因产物,或定位于同一个细胞组分。例如,某个未知的A基因和一些已经被注释到GO数据库中某一生物学过程的B类基因共表达,那么这个未知的A基因很可能与B类基因一样在同一个生物学过程中发挥功能
- 实用贴—注释数据库介绍之GO、KEGG数据库
NCBI数据库
- ncbi就不过多介绍了
- 我感觉在注释结果中, ncbi的结果是最多的, 同时, 也可能是最不准确的
Swissprot数据库
- Swissprot数据库中的基因是经过功能验证的, 我觉得参考价值应该是最高的
- 正相反, uniprot数据库中的另外一个TrEMBL数据库包含的是没有经过功能验证的基因
- 每个序列条目由核心数据(Core Data)和注释数据(Annotation)组成。核心数据包括序列、参考文献和序列的生物来源,而注释数据则描述了:①蛋白质的功能;②蛋白质的翻译后加工修饰,如糖基化(Carbohydration)、磷酸化(Phosphorylation)、乙酰化(Acetylation)、GPI锚定(GPI-anchor)等;③结构域( Domains)和结合位点(Binding Sites),如钙结合区(Calcium Binding Regions)、ATP结合位点(ATP-Binding Sites)、锌指结构域(Zinc Fingers)、同源盒(Homeobox)、Kringle等;④二级结构,四级结构; ⑤和其他蛋白的序列相似性(Sequence Similarity);⑥相关疾病(Associated Diseases)和序列变异( Variants)等 (来自付强)
KEGG数据库
- KEGG数据库是一个有助于了解高级功能和生物学系统的工具。实际上,KEGG通过对生物学过程进行计算机化处理,构建模块并绘制图表,从而对基因的功能进行系统化的分析。KEGG也是将基因组中的一系列基因用一个细胞内的分子相互作用网络连接起来的过程,如一个通路或是一个复合物,通过他们来展现更高一级的生物学功能。目的是从基因组和分子水平上了解高一级层次的功能和与之作用的生物信息资源。KEGG数据库中还包含疾病和药物信息等
InterProScan数据库
- 通过蛋白质结构域和功能位点数据库预测蛋白质功能。是EBI开发的一个集成了蛋白质家族、结构域和功能位点的非冗余数据库。Interproscan整合了一些使用最普及的一些数据库,并应用于功能未知的蛋白进行Interpro注释和GO注释 (来自陈连福的生信博客)
Pfam
- Pfam数据库是蛋白质家族的大集合,每个蛋白质家族由多序列比对 和隐马尔可夫模型(HMM)表示
- 蛋白质通常由一个或多个功能区组成,通常称为结构域。不同的结构域组合产生了天然存在的各种蛋白质。因此,鉴定蛋白质内发生的结构域可以提供对其功能的见解
方法一: 以KEGG的注释结果为主, 筛选出每个品种包含的特异通路及基因
文件准备
- https://www.genome.jp/kegg/catalog/org_list.html下载所有植物通路及包含基因的文件
- 目的区段功能注释文件
基本思路
- 将植物通路构建了一个字典dic1,通路名称为key,包含的基因为value
- 9个文件,每个建一个字典dic2(以一个为例),geneID为key,KEGG注释部分为value
- 取dic2 value中的KEGG_ID到dic1 value中遍历, 匹配成功后,将此 dic1 value对应的key作为最终字典dic3的key,将基因ID+‘==>’+dic2 value作为value
- 这个dic3包含了9个文件中含有keggid能匹配到苹果通路中的所有基因
- 从字典中选择只包含唯一一个品种的通路及其value, 认为此value包含的基因即为该品种的特异功能基因
脚本
这个脚本输出部分写的异常垃圾, 当时只是觉得自己能看懂就行了
# -*- encoding:UTF-8 -*-
import re
import os
import pprint
def text_open(file_path):
'''用于一个通路文件和9个特异基因文件的导入'''
try:
text_name = []
for line in open(file_path):
line = line.strip()
text_name.append(line)
return text_name
except:
print "The 'text_open' has a question!"
def pathway_dic(pathway_text):
'''通路文件生成字典,通路名称为key,通路包含的基因为value'''
try:
dic = {}
partern1 = re.compile('^C')
partern2 = re.compile('^D')
for line in pathway_text:
if partern1.search(line):
key_name =line
dic[key_name] = []
continue
if partern2.search(line):
dic[key_name].append(line)
return dic
except:
print "The 'pathway_dic' has a question!"
def geneid2keggid_dic(pear_text):
'''特异基因信息生成字典1,基因id为key,kegg id为value'''
try:
dic = {}
partern1 = re.compile('K\d{5}')
for line in pear_text:
line_table = line.split('\t')
if partern1.search(line_table[8]):
dic[line_table[0]] = partern1.search(line_table[8]).group()
return dic
except:
print "The 'geneid2keggid_dic' has a question!"
def geneid2keggline_dic(pear_text):
'''特异基因信息生成字典2,基因id为key,kegg注释信息为value'''
try:
dic = {}
partern1 = re.compile('K\d{5}')
for line in pear_text:
line_table = line.split('\t')
if partern1.search(line_table[8]):
dic[line_table[0]] = line_table[8]
return dic
except:
print "The 'geneid2keggline_dic' has q question!"
def nine_list(osdir):
'''将9个文件的列表写入一个更大列表中'''
try:
specific_genefile_list = []
big_table = []
partern1 = re.compile('^P\w+_.*')
# partern1 = re.compile('^sun.*')
for lists in os.listdir(osdir):
if partern1.search(lists):
specific_genefile_list.append(lists)
for file in specific_genefile_list:
big_table.append(text_open(osdir+'/'+file))
return big_table
except:
print "The 'nine_list' has a question!"
def our_assume1(big_dic):
try:
new_dic = {}
for big_dic_key in big_dic.keys():
if big_dic[big_dic_key] != []:
n = 1
small_table = big_dic[big_dic_key][-1].split('==>')
partern2 = re.compile('^P[a-zA-Z]+')
word = partern2.search(small_table[0]).group()
partern1 = re.compile(word)
for big_dic_value in big_dic[big_dic_key]:
if not partern1.search(big_dic_value):
n +=1
if n == 1:
new_dic[big_dic_key] = big_dic[big_dic_key]
return new_dic
except:
print "The 'our_assume1' has a question!"
def our_assume2and3(big_dic):
try:
new_dic1 = {}
new_dic2 = {}
new_dic3 = {}
new_dic4 = {}
for big_dic_key in big_dic.keys():
if big_dic[big_dic_key] != []:
words = []
for big_dic_value in big_dic[big_dic_key]:
small_table = big_dic_value.split('==>')
partern2 = re.compile('^P[a-zA-Z]+')
words.append(partern2.search(small_table[0]).group())
if 'Pbr' in words and 'Ppc' in words and 'Puc' in words and 'Pco' in words and 'Psi' in words and 'Ppw' not in words and 'Puw' not in words and 'Ppy' not in words and 'Pbe' not in words:
new_dic1[big_dic_key] = big_dic[big_dic_key]
if 'Pbr' not in words and 'Ppc' not in words and 'Puc' not in words and 'Pco' not in words and 'Psi' not in words and 'Ppw' in words and 'Puw' in words and 'Ppy' in words and 'Pbe' in words:
new_dic2[big_dic_key] = big_dic[big_dic_key]
if 'Pbr' in words and 'Ppc' in words and 'Puc' in words and 'Ppw' in words and 'Psi' in words and 'Puw' in words and 'Pbe' in words and 'Pco' not in words and 'Ppy' not in words:
new_dic3[big_dic_key] = big_dic[big_dic_key]
if 'Pbr'not in words and 'Ppc'not in words and 'Puc' not in words and 'Ppw'not in words and 'Psi'not in words and 'Puw'not in words and 'Pbe' not in words and 'Pco' in words and 'Ppy' in words:
new_dic4[big_dic_key] = big_dic[big_dic_key]
return new_dic1, new_dic2, new_dic3, new_dic4
except:
print "The 'our_assume2' has a question!"
if __name__ == '__main__':
'''problems 不能找到其他物种没有定义的通路;没有KEGG注释的基因没办法参与定位;没有定位到的基因可能位于非特异区域'''
big_table = nine_list('/Users/suncongrui/home/03_workshop/specific_gene/target_gene')
pathway_text = text_open('/Users/suncongrui/home/03_workshop/specific_gene/plant/ko00001.keg')
pathway_dic1 = pathway_dic(pathway_text)
big_dic = {}
for def2_key in pathway_dic1.keys():
big_dic[def2_key] = []
for specise in big_table:
geneid2keggline_dic1 = geneid2keggline_dic(specise)
geneid2keggid_dic1 = geneid2keggid_dic(specise)
for geneid in geneid2keggid_dic1.keys():
partern1 = re.compile(geneid2keggid_dic1[geneid])
for pathway_dir_key in pathway_dic1.keys():
for pathway_dir_value in pathway_dic1[pathway_dir_key]:
if partern1.search(pathway_dir_value):
big_dic[pathway_dir_key].append(geneid+'==>'+geneid2keggline_dic1[geneid])
sun = our_assume1(big_dic)
cong1, cong2, rui1, rui2 = our_assume2and3(big_dic)
pp = pprint.PrettyPrinter(indent=4)
# pp.pprint(sun)
# pp.pprint(cong1)
# pp.pprint(cong2)
# pp.pprint(rui1)
pp.pprint(rui2)
部分结果展示
输出异常垃圾...

垃圾输出
此方法的问题
- 9个品种的很多基因没有KEGG注释结果
- 有的基因在很多通路中起作用, 但可能有主次之分, 这样的基因也过滤掉了
- 没有定位到的基因可能位于非特异区域
方法二: 利用pfam的注释结果, 过滤掉每个品种具有相同结构域的基因, 对于剩下的基因, 利用其swissprot id 在DAVID数据库中查找他们的功能信息, 单个进行筛选
文件准备
- 目的区段功能注释文件
wget ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
脚本
- 脚本一: 过滤掉相同结构域的基因
# -*- encoding:UTF-8 -*-
import re
import os
def text_open(file_path):
'''用于一个通路文件和9个特异基因文件的导入'''
try:
text_name = []
for line in open(file_path):
line = line.strip()
text_name.append(line)
return text_name
except:
print "The 'text_open' has a question!"
def nine_list(osdir):
'''将9个文件的id-pfam列写入一个更大列表中'''
try:
specific_genefile_list = []
big_table = []
partern1 = re.compile('^P\w+_.*')
for lists in os.listdir(osdir):
if partern1.search(lists):
specific_genefile_list.append(lists)
for file in specific_genefile_list:
pfam = []
text_name = text_open(osdir+'/'+file)
for line in text_name:
line_table = line.split('\t')
pfam.append(line_table[0]+'\t'+line_table[-1])
big_table.append(pfam)
return big_table
except:
print "The 'nine_list' has a question!"
if __name__ == '__main__':
big_table = nine_list('/Users/suncongrui/home/03_workshop/specific_gene/target_gene')
table = []
for i in range(9):
big_table_copy = big_table[:]
table1 = []
table2 = []
del big_table_copy[i]
for small_table_copy in big_table_copy:
for line in small_table_copy:
line_table1 = line.split('\t')
table1.append(line_table1[1])
for line in big_table[i]:
line_table2 = line.split('\t')
if line_table2[1] in table1:
table2.append(line_table2[0]+'\t'+line_table2[1]+'\t'+'NO')
else:
table2.append(line_table2[0]+'\t'+line_table2[1]+'\t'+'YES')
table.append(table2)
for b in table:
for c in b:
print c
- 脚本二: 因为swissprot注释结果没有id, 所有用下载的数据库匹配对应基因的id
# -*- encoding:UTF-8 -*-
import re
import os
def text_open(file_path):
'''用于一个通路文件和9个特异基因文件的导入'''
try:
text_name = []
for line in open(file_path):
line = line.strip()
text_name.append(line)
return text_name
except:
print "The 'text_open' has a question!"
def nine_list(osdir):
'''将9个文件的id-swissprot-id列写入一个更大列表中'''
try:
specific_genefile_list = []
big_table = []
partern1 = re.compile('^P\w+_.*')
for lists in os.listdir(osdir):
if partern1.search(lists):
specific_genefile_list.append(lists)
for file in specific_genefile_list:
text_name = text_open(osdir+'/'+file)
for line in text_name:
line_table = line.split('\t')
big_table.append(line_table[0]+'\t'+line_table[7])
return big_table
except:
print "The 'nine_list' has a question!"
if __name__ == '__main__':
big_table = nine_list('/home/Dai_XG/software/user/74/target_gene')
swissprot_text = text_open('/home/Dai_XG/software/user/74/uniprot_sprot.head')
table = []
for element1 in big_table:
line_table = element1.split('\t')
n = 0
for element2 in swissprot_text:
if line_table[1] == 'NA':
continue
if line_table[1] in element2:
split = element2.split('|')
print line_table[0]+'\t'+split[1]+'\t'+line_table[1]
n += 1
if n == 0:
print line_table[0]+'\t'+'NO'+'\t'+line_table[1]
查找部分核心展示
这里要感谢一下chuxinxzz同学的帮助, 效率提高了不少
- 将运行脚本的到的结果id写入单独的txt文件中
- 提交到DAVID数据库中, 可以得到比较全面的功能列表
-
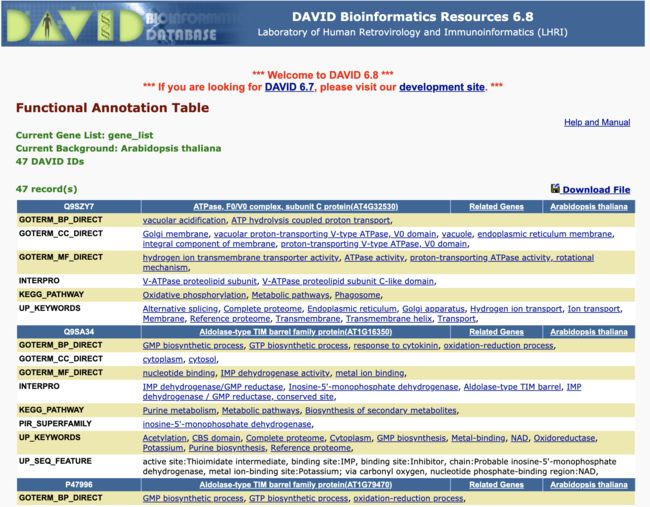 DAVID
DAVID - 然后挨着往后读, 找到疑似结果就记录一下, 这比单个找出来去每个数据库搜索方便了不少
最后
夏天很热 -_-

热死我了