NGS分析流程
NGS实验步骤
核酸提取与检测、文库构建与文库检测、上机测序
生信分析步骤
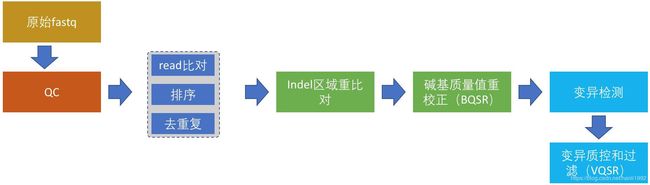
1. 质量分析
fastqc、multiqc、SolexaQA
测序数据的质量好坏会影响我们的下游分析。但不同的测序平台其测序错误率的图谱都是有差别的。因此,非常建议在我们分析测序数据之前先搞清楚如下两个地方:
-
原始数据是通过哪种测序平台产生的,它们的错误率分布是怎么样的,是否有一定的偏向性和局限性,是否会显著受GC含量的影响等;
-
评估它们有可能影响哪些方面的分析;
2. 数据过滤
去除接头序列和低质量序列,常用工具:SOAPnuke、cutadapt、untrimmed、fastp、sickle、seqtk、Trimmomatic、Trim Galore(合并了FastQC和cutadapt)
3. 比对
使用BWA(exact match)软件把这些短序列和参考基因组进行对比, 确定短序列在基因组上的位置,生成*.sam文件。
4. 排序
用samtools等进行比对、排序、建索引,得到reads在参考序列中的位置及质量值,并进行数据格式的转换,得到*.bam文件。
5. Bam文件再处理
remove duplicates → indel realign → BQSR
remove duplicates
使用Picard软件把测序产生的冗余信息和噪声去掉,并对数据质量进行评价。
在制备文库的过程中,由于PCR扩增过程中会存在一些偏差,也就是说有的序列会被过量扩增。这样,在比对的时候,这些过量扩增出来的完全相同的序列就会比对到基因组的相同位置。而这些过量扩增的reads并不是基因组自身固有序列,不能作为变异检测的证据,因此,要尽量去除这些由PCR扩增所形成的duplicates,这一步可以使用picard-tools来完成。去重复的过程是给这些序列设置一个flag以标志它们,方便GATK的识别。还可以设置 REMOVE_DUPLICATES=true 来丢弃duplicated序列。对于是否选择标记或者删除,对结果应该没有什么影响,GATK官方流程里面给出的例子是仅做标记不删除。这里定义的重复序列是这样的:如果两条reads具有相同的长度而且比对到了基因组的同一位置,那么就认为这样的reads是由PCR扩增而来,就会被GATK标记。
indel realign
BWA采取exact match策略,在indel附近比对效果不好,需进行局部重新比对。一般来说,绝大部分需要进行重新比对的基因组区域,都是因为插入/缺失的存在,因为在indel附近的比对会出现大量的碱基错配,这些碱基的错配很容易被误认为SNP。还有,在比对过程中,比对算法对于每一条read的处理都是独立的,不可能同时把多条reads与参考基因组比对来排错。因此,即使有一些reads能够正确的比对到indel,但那些恰恰比对到indel开始或者结束位置的read也会有很高的比对错误率,这都是需要重新比对的。Local realignment就是将由indel导致错配的区域进行重新比对,将indel附近的比对错误率降到最低。可以使用GATK工具进行indel重新比对。
BQSR(Base Quality Score Recalibration)
BQSR是对bam文件里reads的碱基质量值进行重新校正,使最后输出的bam文件中reads中碱基的质量值能够更加接近真实的与参考基因组之间错配的概率。例如,在reads碱基质量值被校正之前,我们要保留质量值在Q25以上的碱基,但是实际上质量值在Q25的这些碱基的错误率在1%,也就是说质量值只有Q20,这样就会对后续的变异检测的可信度造成影响。还有,在边合成边测序的测序过程中,在reads末端碱基的错误率往往要比起始部位更高。另外,AC的质量值往往要低于TG。BQSR的就是要对这些质量值进行校正。使用GATK对重新比对的BAM文件做质量校准。
6. 获取突变
用GATK等得到.vcf文件,从中获取如外显子区域的数据,错义突变,热点突变,靶向药物相关的突变等感兴趣的突变信息。
7. 注释
使用Annovar对这些变异位点进行功能注释, 得到一个易于理解的变异位点列表。
| 相关格式 |
名称 |
类型 |
常见后缀 |
| 序列与质量分数 |
FASTA |
文本 |
.fa/.fna/.fasta |
| FASTAQ |
文本 |
.fq/.fastq |
|
| 序列比对 |
SAM |
文本 |
.sam |
| BAM |
二进制 |
.bam |
|
| 序列组装 |
ACE |
文本 |
.ace |
| AFG |
文本 |
.afg |
|
| CAF |
文本 |
.caf |
|
| 突变 |
VCF |
文本 |
.vcf |
| 序列注释及可视化 |
BED |
文本 |
.bed |
Fasta
一种基于文本用于表示核苷酸序列或氨基酸序列的格式(.fa, .fasta, .fna)
每一条序列的第一行以“>”开头,而跟随“>”的是序列的ID号(即唯一的标识符)及对该序列的描述信息;
第二行开始是序列内容,序列短于61nt的,则一行排列完;序列长于61nt的,则每行存储61nt,最后剩下小于61nt的,在最后一行排列完;第二条序列另起一行,仍然由“>”和序列的ID号开始,以此类推。

Fastq
Fastq是基于文本的保存生物序列(通常是核酸序列)和其测序质量信息的标准格式(.fastq, .fq)。
第一行以“@”符号开头,后面紧跟一个序列的描述信息;
第二行是该序列的内容;
第三行以“+”符号开头,后面可以是该序列的描述信息,也可省略;
第四行是第二行中的序列内容每个碱基所对应的测序质量值。
这里我们假定碱基的测序错误率为p_error,质量值为Q,它们之间的关系如下:
Q = -10log(p_error)

SAM
SAM/BAM格式专用于存储基于参考序列的比对序列,SAM(Sequence Alignment Map)是“序列比对映射”的首字母缩写,是带有比对信息的序列文件(即告诉你这个reads在染色体上的位置等),用于储存序列数据。
下图表示read和参考基因组比对可能出现的情况,

Coor:坐标的简写,方便查看比对
ref:参考序列
r001/2表示paired end数据,
r003是嵌合read,
r004则是原序列打断后比对结果。
经过专门的比对软件,如BWA、BOWTIE2等,得到的SAM文件如下所示,需要研究的就是如下这几行。

第一部分:SAM Header(非强制)
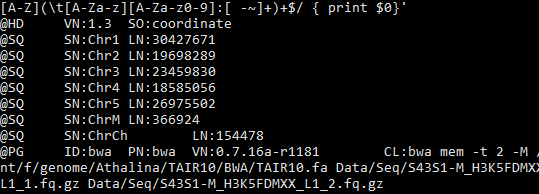
@CO,任意的说明信息
@HD表示参考基因组的排序情况。
@PG记录运行的命令,以便你检查代码。对于GATK还需要提供@RG给出每个read所在group的信息,只要保证是独一即可。
@RG,比对上的序列(read)说明
@SQ是参考基因组的每一条序列的具体信息,命名和长度。
第二部分:联配必要信息
每一行包括十一项,通过Tab键分隔。
第一列:read name,read的名字通常包括测序平台等信息
eg.ILLUMINA-379DBF:1:1:3445:946#0/1
第二列:sum of flags,为flag的总和(整数),
eg.16
flag取值如下:
1: 代表这个序列采用的是PE双端测序
2: 代表这个序列和参考序列完全匹配,没有错配和插入缺失
4: 代表这个序列没有mapping到参考序列上
8: 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上
16:代表这个序列比对到参考序列的负链上
32:代表这个序列对应的另一端序列比对到参考序列的负链上
64: 代表这个序列是R1端序列, read1;
128: 代表这个序列是R2端序列,read2;
256: 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的
512: 代表这个序列在QC时失败了,被过滤不掉了(# 这个标签不常用)
1024: 代表这个序列是PCR重复序列(#这个标签不常用)
2048: 代表这个序列是补充的比对(#这个标签具体什么意思,没搞清楚,但是不常用)
上面的这几个标签都是2的n次方,这样的数列有一个特点,就是随机挑选其中的几个,它们的和是唯一的,比如65 只能是1 和 64 组成,代表这个序列是双端测序,而且是read1假如说标记为以上列举出的数目,就可以直接推断出匹配的情况。假如说标记不是以上列举出的数字,比如说83=(64+16+2+1),就是这几种情况值和。
第三列:RNAME,比对到参考序列上的染色体号。若是无法比对,则是*
eg.chr1
第四列:position,read比对到参考序列上,第一个碱基所在的位置。若是无法比对,则是0
eg.36576599
第五列:Mapping quality,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一。
eg.42
第六列:CIGAR值,碱基匹配上的碱基数。
eg. 37M1D2M1I,这段字符的意思是37个匹配,1个参考序列上的删除,2个匹配
#M match/mismatch
#I insertion
#D deletion
#extended cigar
#N gap
#S substitution
#H hard clipping
#P padding
#= sequence match
#X sequence mismatch
注:第七列到第九列是mate(备注1)的信息,若是单末端测序这几列均无意义。
第七列:mate序列所在参考序列的名称,实际上就是比对到的染色体号,若是没有mate,则是*,同一个片段,用'='
eg.*
第八列:mate position,mate比对到参考序列上的第一个碱基位置,若无mate,则为0
eg.0
第九列:估计出的片段的长度,当mate序列位于本序列上游时该值为负值。Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0;
eg.0
第十列:Sequence,就是read的碱基序列,如果不存储此类信息,此处为'*',注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;如果是比对到互补链上则对read进行了reverse completed
eg.CGTTTCTGTGGGTGATGGGCCTGAGGGGCGTTCTCN
第十一列:ASCII,read质量的ASCII编码。
eg.PY[[YY_______________QQQQbILKIGEFGKB
第三部分:可选信息
除了之前的11列必须要有的信息外,后面的其他列都是不同的比对软件自定义的额外信息,称之为标签(TAG)。
AS:i 匹配的得分
XS:i 第二好的匹配的得分
YS:i mate 序列匹配的得分
XN:i 在参考序列上模糊碱基的个数
XM:i 错配的个数
XO:i gap open的个数
XG:i gap 延伸的个数
NM:i 经过编辑的序列
YF:i 说明为什么这个序列被过滤的字符串
YT:Z
MD:Z 代表序列和参考序列错配的字符串
BAM
BAM是(SAM的)二进制格式,因为它是压缩的、所以数据量更小;因为它是有索引的,所以可以更快地访问它。
VCF(Variant Call Format)
| #CHROM |
POS |
ID |
REF |
ALT |
QUAL |
FILTER |
INFO |
FORMAT |
Sample1 |
VCF文件分为两部分内容:以“#”开头的注释部分;没有“#”开头的主体部分。
CHROM和POS:代表参考序列名和variant的位置;如果是INDEL的话,位置是INDEL的第一个碱基位置。
ID:variant的ID。比如在dbSNP中有该SNP的id,则会在此行给出;若没有,则用’.'表示其为一个novel variant。
REF 和 ALT:参考序列的碱基 和 Variant的碱基。
QUAL:Phred格式(Phred_scaled)的质量值,表 示在该位点存在variant的可能性;该值越高,则variant的可能性越大;计算方法:Phred值 = -10 * log (1-p) p为variant存在的概率; 通过计算公式可以看出值为10的表示错误概率为0.1,该位点为variant的概率为90%。
FILTER:使用上一个QUAL值来进行过滤的话,是不够的。GATK能使用其它的方法来进行过滤,过滤结果中通过则该值为”PASS”;若variant不可靠,则该项不为”PASS”或”.”。
INFO: 这一行是variant的详细信息。
FORMAT 和 NA12878:这两行合起来提供了’NA12878′这个sample的基因型的信息。’NA12878′代表这该名称的样品,是由BAM文件中的@RG下的 SM 标签决定的。
GT:样品的基因型(genotype)。两个数字中间用’/'分 开,这两个数字表示双倍体的sample的基因型。0 表示样品中有ref的allele; 1 表示样品中variant的allele; 2表示有第二个variant的allele。因此: 0/0 表示sample中该位点为纯合的,和ref一致; 0/1 表示sample中该位点为杂合的,有ref和variant两个基因型; 1/1 表示sample中该位点为纯合的,和variant一致。
AD 和 DP:AD(Allele Depth)为sample中每一种allele的reads覆盖度,在diploid中则是用逗号分割的两个值,前者对应ref基因型,后者对应variant基因型; DP(Depth)为sample中该位点的覆盖度。
GQ:基因型的质量值(Genotype Quality)。Phred格式(Phred_scaled)的质量值,表示在该位点该基因型存在的可能性;该值越高,则Genotype的可能性越 大;计算方法:Phred值 = -10 * log (1-p) p为基因型存在的概率。
PL:指定的三种基因型的质量值(provieds the likelihoods of the given genotypes)。这三种指定的基因型为(0/0,0/1,1/1),这三种基因型的概率总和为1。和之前不一致,该值越大,表明为该种基因型的可能 性越小。 Phred值 = -10 * log (p) p为基因型存在的概率。