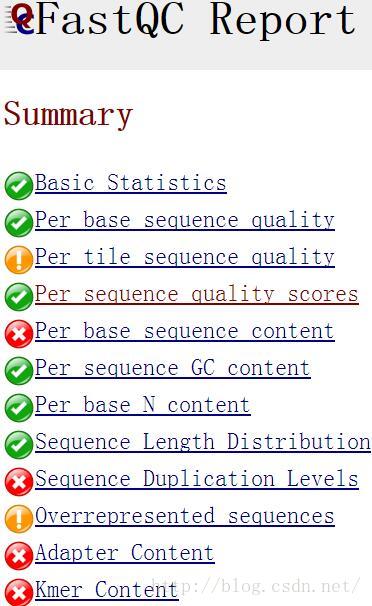
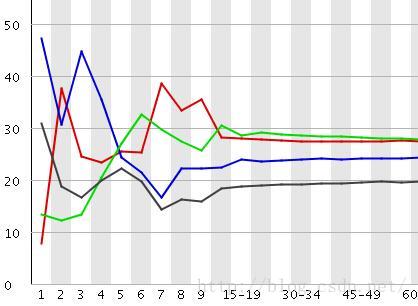
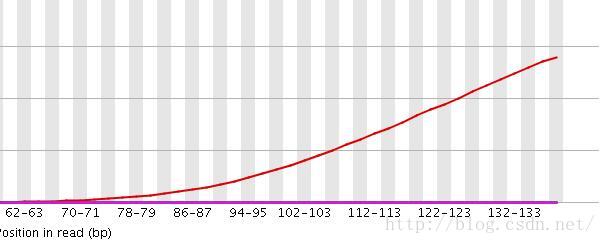
- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- android系统selinux中添加新属性property
辉色投像
1.定位/android/system/sepolicy/private/property_contexts声明属性开头:persist.charge声明属性类型:u:object_r:system_prop:s0图12.定位到android/system/sepolicy/public/domain.te删除neverallow{domain-init}default_prop:property
- 铭刻于星(四十二)
随风至
69夜晚,绍敏同学做完功课后,看了眼房外,没听到动静才敢从书包的夹层里拿出那个心形纸团。折痕压得很深,都有些旧了,想来是已经写好很久了。绍敏同学慢慢地、轻轻地捏开折叠处,待到全部拆开后,又反复抚平纸张,然后仔细地一字字默看。只是开头的三个字是第一次看到,让她心漏跳了几拍。“亲爱的绍敏:从四年级的时候,我就喜欢你了,但是我一直不敢说,怕影响你学习。六年级的时候听说有人跟你表白,你接受了,我很难过,但
- 底层逆袭到底有多难,不甘平凡的你准备好了吗?让吴起给你说说
造命者说
底层逆袭到底有多难,不甘平凡的你准备好了吗?让吴起给你说说我叫吴起,生于公元前440年的战国初期,正是群雄并起、天下纷争不断的时候。后人说我是军事家、政治家、改革家,是兵家代表人物。评价我一生历仕鲁、魏、楚三国,通晓兵家、法家、儒家三家思想,在内政军事上都有极高的成就。周安王二十一年(公元前381年),因变法得罪守旧贵族,被人乱箭射死。我出生在卫国一个“家累万金”的富有家庭,从年轻时候起就不甘平凡
- 想家
爆米花机
也许不同于大家对家乡的思念,我对家乡甚至是疯狂的不舍。还未踏出车站就感觉到幸福,我享受这里的夕阳、这里的浓烈柴火味、这里每一口家常菜。我是宅女,我贪恋家的安逸。刚刚踏出大学校门,初出茅庐,无法适应每年只能国庆和春节回家。我焦虑、失眠、无端发脾气,是无法适应工作的节奏,是无法接受我将一步步离开家乡的事实。我不想承认自己胸无大志,选择再次踏上征程。图片发自App
- 【iOS】MVC设计模式
Magnetic_h
iosmvc设计模式objective-c学习ui
MVC前言如何设计一个程序的结构,这是一门专门的学问,叫做"架构模式"(architecturalpattern),属于编程的方法论。MVC模式就是架构模式的一种。它是Apple官方推荐的App开发架构,也是一般开发者最先遇到、最经典的架构。MVC各层controller层Controller/ViewController/VC(控制器)负责协调Model和View,处理大部分逻辑它将数据从Mod
- element实现动态路由+面包屑
软件技术NINI
vue案例vue.js前端
el-breadcrumb是ElementUI组件库中的一个面包屑导航组件,它用于显示当前页面的路径,帮助用户快速理解和导航到应用的各个部分。在Vue.js项目中,如果你已经安装了ElementUI,就可以很方便地使用el-breadcrumb组件。以下是一个基本的使用示例:安装ElementUI(如果你还没有安装的话):你可以通过npm或yarn来安装ElementUI。bash复制代码npmi
- 2021年12月19日,春蕾教育集团团建活动感受——黄晓丹
黄错错加油
感受:1.从陌生到熟悉的过程。游戏环节让我们在轻松的氛围中得到了锻炼,也增长了不少知识。2.游戏过程中,我们贡献的是个人力量,展现的是团队的力量。它磨合的往往不止是工作的熟悉,更是观念上契合度的贴近。3.这和工作是一样的道理。在各自的岗位上,每个人摆正自己的位置、各司其职充分发挥才能,并团结一致劲往一处使,才能实现最大的成功。新知:1.团队精神需要不断地创新。过去,人们把创新看作是冒风险,现在人们
- 爬山后遗症
璃绛
爬山,攀登,一步一步走向制高点,是一种挑战。成功抵达是一种无法言语的快乐,在山顶吹吹风,看看风景,这是从未有过的体验。然而,爬山一时爽,下山腿打颤,颠簸的路,一路向下走,腿部力量不够,走起来抖到不行,停不下来了!第二天必定腿疼,浑身酸痛,坐立难安!
- 《策划经理回忆录之二》
路基雅虎
话说三年变六年,飘了,飘了……眨眼,2013年5月,老吴回到了他的家乡——油城从新开启他的工作幻想症生涯。很庆幸,这是一家很有追求,同时敢于尝试的,且实力不容低调的新星房企——金源置业(前身泰源置业)更值得庆幸的是第一个盘就是油城十路的标杆之一:金源盛世。2013年5月,到2015年11月,两年的陪伴,迎来了一场大爆发。2000个筹,5万/筹,直接回笼1个亿!!!这……让我开始认真审视这座看似五线
- Long类型前后端数据不一致
igotyback
前端
响应给前端的数据浏览器控制台中response中看到的Long类型的数据是正常的到前端数据不一致前后端数据类型不匹配是一个常见问题,尤其是当后端使用Java的Long类型(64位)与前端JavaScript的Number类型(最大安全整数为2^53-1,即16位)进行数据交互时,很容易出现精度丢失的问题。这是因为JavaScript中的Number类型无法安全地表示超过16位的整数。为了解决这个问
- 30天风格练习-DAY2
黄希夷
Day2(重义)在一个周日/一周的最后一天,我来到位于市中心/市区繁华地带的一家购物中心/商场,中心内人很多/熙熙攘攘。我注意到/看见一个独行/孤身一人的年轻女孩/,留着一头引人注目/长过腰际的头发,上身穿一件暗红色/比正红色更深的衣服/穿在身体上的东西。走下扶梯的时候,她摔倒了/跌向地面,在她正要站起来/让身体离开地面的时候,过长/超过一般人长度的头发被支撑身体/躯干的手掌压/按在下面,她赶紧用
- 消息中间件有哪些常见类型
xmh-sxh-1314
java
消息中间件根据其设计理念和用途,可以大致分为以下几种常见类型:点对点消息队列(Point-to-PointMessagingQueues):在这种模型中,消息被发送到特定的队列中,消费者从队列中取出并处理消息。队列中的消息只能被一个消费者消费,消费后即被删除。常见的实现包括IBM的MQSeries、RabbitMQ的部分使用场景等。适用于任务分发、负载均衡等场景。发布/订阅消息模型(Pub/Sub
- 三大师传
beca酱
巴尔扎克的作品被誉为“法国社会的一面镜子”。文学大师维克多·雨果对巴尔扎克的评价是:“在最伟大的人物中间,巴尔扎克是名列前茅者;在最优秀的人物中间,巴尔扎克是佼佼者之一。”一个原本寂寂无名的小人物,从地中海的某个海岛上,只身一人来到巴黎,没有朋友,也没有名望。作为一个一文不名的外乡人,凭着赤手空拳赢得了巴黎,征服了整个法兰西,并且赢得了世界。这个人就是十九世纪法国伟大的军事家、政治家,法兰西第一帝
- 开心
蒋泳频
从无比抗拒来上课到接受,感动,收获~看着波哥成长,晶晶幸福笑容满面。感觉自己做的事情很有意义,很开心!还有3个感召目标就是还有三个有缘人,哈哈。明天感召去明日计划:8:30-11:00小公益11:00-21点上班,感召图片发自App图片发自App图片发自App
- 回溯 Leetcode 332 重新安排行程
mmaerd
Leetcode刷题学习记录leetcode算法职场和发展
重新安排行程Leetcode332学习记录自代码随想录给你一份航线列表tickets,其中tickets[i]=[fromi,toi]表示飞机出发和降落的机场地点。请你对该行程进行重新规划排序。所有这些机票都属于一个从JFK(肯尼迪国际机场)出发的先生,所以该行程必须从JFK开始。如果存在多种有效的行程,请你按字典排序返回最小的行程组合。例如,行程[“JFK”,“LGA”]与[“JFK”,“LGB
- Python数据分析与可视化实战指南
William数据分析
pythonpython数据
在数据驱动的时代,Python因其简洁的语法、强大的库生态系统以及活跃的社区,成为了数据分析与可视化的首选语言。本文将通过一个详细的案例,带领大家学习如何使用Python进行数据分析,并通过可视化来直观呈现分析结果。一、环境准备1.1安装必要库在开始数据分析和可视化之前,我们需要安装一些常用的库。主要包括pandas、numpy、matplotlib和seaborn等。这些库分别用于数据处理、数学
- 每日一题——第八十四题
互联网打工人no1
C语言程序设计每日一练c语言
题目:编写函数1、输入10个职工的姓名和职工号2、按照职工由大到小顺序排列,姓名顺序也随之调整3、要求输入一个职工号,用折半查找法找出该职工的姓名#define_CRT_SECURE_NO_WARNINGS#include#include#defineMAX_EMPLOYEES10typedefstruct{intid;charname[50];}Empolyee;voidinputEmploye
- 每日一题——第八十二题
互联网打工人no1
C语言程序设计每日一练c语言
题目:将一个控制台输入的字符串中的所有元音字母复制到另一字符串中#include#include#include#include#defineMAX_INPUT1024boolisVowel(charp);intmain(){charinput[MAX_INPUT];charoutput[MAX_INPUT];printf("请输入一串字符串:\n");fgets(input,sizeof(inp
- 《庄子.达生9》
钱江潮369
【原文】孔子观于吕梁,县水三十仞,流沫四十里,鼋鼍鱼鳖之所不能游也。见一丈夫游之,以为有苦而欲死也,使弟子并流而拯之。数百步而出,被发行歌而游于塘下。孔子从而问焉,曰:“吾以子为鬼,察子则人也。请问,‘蹈水有道乎’”曰:“亡,吾无道。吾始乎故,长乎性,成乎命。与齐俱入,与汩偕出,从水之道而不为私焉。此吾所以蹈之也。”孔子曰:“何谓始乎故,长乎性,成乎命?”曰:“吾生于陵而安于陵,故也;长于水而安于
- Pyecharts数据可视化大屏:打造沉浸式数据分析体验
我的运维人生
信息可视化数据分析数据挖掘运维开发技术共享
Pyecharts数据可视化大屏:打造沉浸式数据分析体验在当今这个数据驱动的时代,如何将海量数据以直观、生动的方式展现出来,成为了数据分析师和企业决策者关注的焦点。Pyecharts,作为一款基于Python的开源数据可视化库,凭借其丰富的图表类型、灵活的配置选项以及高度的定制化能力,成为了构建数据可视化大屏的理想选择。本文将深入探讨如何利用Pyecharts打造数据可视化大屏,并通过实际代码案例
- 第四天旅游线路预览——从换乘中心到喀纳斯湖
陟彼高冈yu
基于Googleearthstudio的旅游规划和预览旅游
第四天:从贾登峪到喀纳斯风景区入口,晚上住宿贾登峪;换乘中心有4路车,喀纳斯①号车,去喀纳斯湖,路程时长约5分钟;将上面的的行程安排进行动态展示,具体步骤见”Googleearthstudio进行动态轨迹显示制作过程“、“Googleearthstudio入门教程”和“Googleearthstudio进阶教程“相关内容,得到行程如下所示:Day4-2-480p
- Python教程:一文了解使用Python处理XPath
旦莫
Python进阶python开发语言
目录1.环境准备1.1安装lxml1.2验证安装2.XPath基础2.1什么是XPath?2.2XPath语法2.3示例XML文档3.使用lxml解析XML3.1解析XML文档3.2查看解析结果4.XPath查询4.1基本路径查询4.2使用属性查询4.3查询多个节点5.XPath的高级用法5.1使用逻辑运算符5.2使用函数6.实战案例6.1从网页抓取数据6.1.1安装Requests库6.1.2代
- 从鸡肉高汤到记忆的魔法再到有效提示的艺术
步子哥
人工智能
还记得小时候那些天马行空的白日梦吗?也许只要按下键盘上的某个神奇组合,电脑就会发出滴滴的声响,一个隐藏的世界突然在你眼前展开,让你获得超凡的能力,摆脱平凡的生活。这听起来像是玩过太多电子游戏的幻想,但实际上,间隔重复系统给人的感觉惊人地相似。在最佳状态下,这些系统就像魔法一样神奇。本文将以一个看似平凡的鸡肉高汤食谱为例,深入浅出地探讨如何编写有效的间隔重复提示,让你像掌握烹饪技巧一样轻松地掌握记忆
- 谁家酒器最绝唱,藏在酒厂人未知?景阳冈酒厂先秦藏品大揭秘
李虓酒评论
文/王赛时中国的酒器酒具历史久远,举世闻名。从北京的故宫博物院、中国国家博物馆,到世界各国的大型博物馆,都以能够收藏中国古代酒具而夸耀。但很少有人知道,在山东阳谷景阳冈酒厂,默默地收藏了两千件中国酒器。这些酒器,就封藏在景阳冈的酒道馆里。其中有一些青铜酒器,一睡就是三、四千年,堪称无声国宝,堪作无字史书!今天,我将引领诸位首先窥视一下景阳冈酒道馆的9件先秦藏品,你自己来说震撼不震撼。提示:这只是景
- 下载github patch到本地
小米人er
我的博客gitpatch
以下是几种从GitHub上下载以.patch结尾的补丁文件的方法:通过浏览器直接下载打开包含该.patch文件的GitHub仓库。在仓库的文件列表中找到对应的.patch文件。点击该文件,浏览器会显示文件的内容,在页面的右上角通常会有一个“Raw”按钮,点击它可以获取原始文件内容。然后在浏览器中使用快捷键(如Ctrl+S或者Command+S)将原始文件保存到本地,选择保存的文件名并确保后缀为.p
- 18-115 一切思考不能有效转化为行动,都TM是扯淡!
成长时间线
7月25号写了一篇关于为什么会断更如此严重的反思,然而,之后日更仅仅维持了一周,又出现了这次更严重的现象。从8月2号到昨天8月6号,5天!又是5天没有更文!虽然这次断更时间和上次一样,那为什么说这次更严重?因为上次之后就分析了问题的原因,以及应该如何解决,按理说应该会好转,然而,没过几天严重断更的现象再次出现,想想,经过反思,问题依然没有解决与改变,这让我有些担忧。到底是哪里出了问题,难道我就真的
- 山东大学小树林支教调研团青青仓木队——翟晓楠
山东大学青青仓木队
过了半年,又一次启程,又一次回到支教的初心之地。比起上一次的试探与不安,我更多了一丝稳重与熟练。心境、处境也都随着半个学期的过去而变得不同,半个学期中,身体上的,心理上的,太多的逆境让我变得步履维艰,曲曲折折,弯弯绕绕,我仿佛打不起精神,没有胃口,没有动力。感觉走的不顺畅的时候,支教这个旅程,给了我力量。自告奋勇承担起队长这一职务的我,从组织时的复杂和困难的经历,协调各种问题,从无到有,和校长和队
- 《中华小厨师》单行VS爱藏:姜是老的辣,书是新的好
cicoky
《汉书·郦食其传》有曰:“王者以民为天,而民以食为天。”自古以来,吃饱饭是每一个人的基本要求,而吃好饭却是每一个人的最终追求。于是,厨师这一职业孕育而生,其渊源之久,甚至可追溯到4000年前的奴隶时代。职业本身无贵贱,但职业能力却有高低之分。所以一家餐馆生意好不好,厨师的水平决定一切,而站在所有厨师顶端的就被称之为“特级厨师”。今天要说的就是一个关于“特级厨师刘昴星”的故事。连载历程1995年第4
- 拥有断舍离的心态,过精简生活--《断舍离》读书笔记
爱吃丸子的小樱桃
不知不觉间房间里的东西越来越多,虽然摆放整齐,但也时常会觉得空间逼仄,令人心生烦闷。抱着断舍离的态度,我开始阅读《断舍离》这本书,希望从书中能找到一些有效的方法,帮助我实现空间、物品上的断舍离。《断舍离》是日本作家山下英子通过自己的经历、思考和实践总结而成的,整体内涵也从刚开始的私人生活哲学的“断舍离”升华成了“人生实践哲学”,接着又成为每个人都能实行的“改变人生的断舍离”,从“哲学”逐渐升华成“
- Js函数返回值
_wy_
jsreturn
一、返回控制与函数结果,语法为:return 表达式;作用: 结束函数执行,返回调用函数,而且把表达式的值作为函数的结果 二、返回控制语法为:return;作用: 结束函数执行,返回调用函数,而且把undefined作为函数的结果 在大多数情况下,为事件处理函数返回false,可以防止默认的事件行为.例如,默认情况下点击一个<a>元素,页面会跳转到该元素href属性
- MySQL 的 char 与 varchar
bylijinnan
mysql
今天发现,create table 时,MySQL 4.1有时会把 char 自动转换成 varchar
测试举例:
CREATE TABLE `varcharLessThan4` (
`lastName` varchar(3)
) ;
mysql> desc varcharLessThan4;
+----------+---------+------+-
- Quartz——TriggerListener和JobListener
eksliang
TriggerListenerJobListenerquartz
转载请出自出处:http://eksliang.iteye.com/blog/2208624 一.概述
listener是一个监听器对象,用于监听scheduler中发生的事件,然后执行相应的操作;你可能已经猜到了,TriggerListeners接受与trigger相关的事件,JobListeners接受与jobs相关的事件。
二.JobListener监听器
j
- oracle层次查询
18289753290
oracle;层次查询;树查询
.oracle层次查询(connect by)
oracle的emp表中包含了一列mgr指出谁是雇员的经理,由于经理也是雇员,所以经理的信息也存储在emp表中。这样emp表就是一个自引用表,表中的mgr列是一个自引用列,它指向emp表中的empno列,mgr表示一个员工的管理者,
select empno,mgr,ename,sal from e
- 通过反射把map中的属性赋值到实体类bean对象中
酷的飞上天空
javaee泛型类型转换
使用过struts2后感觉最方便的就是这个框架能自动把表单的参数赋值到action里面的对象中
但现在主要使用Spring框架的MVC,虽然也有@ModelAttribute可以使用但是明显感觉不方便。
好吧,那就自己再造一个轮子吧。
原理都知道,就是利用反射进行字段的赋值,下面贴代码
主要类如下:
import java.lang.reflect.Field;
imp
- SAP HANA数据存储:传统硬盘的瓶颈问题
蓝儿唯美
HANA
SAPHANA平台有各种各样的应用场景,这也意味着客户的实施方法有许多种选择,关键是如何挑选最适合他们需求的实施方案。
在 《Implementing SAP HANA》这本书中,介绍了SAP平台在现实场景中的运作原理,并给出了实施建议和成功案例供参考。本系列文章节选自《Implementing SAP HANA》,介绍了行存储和列存储的各自特点,以及SAP HANA的数据存储方式如何提升空间压
- Java Socket 多线程实现文件传输
随便小屋
javasocket
高级操作系统作业,让用Socket实现文件传输,有些代码也是在网上找的,写的不好,如果大家能用就用上。
客户端类:
package edu.logic.client;
import java.io.BufferedInputStream;
import java.io.Buffered
- java初学者路径
aijuans
java
学习Java有没有什么捷径?要想学好Java,首先要知道Java的大致分类。自从Sun推出Java以来,就力图使之无所不包,所以Java发展到现在,按应用来分主要分为三大块:J2SE,J2ME和J2EE,这也就是Sun ONE(Open Net Environment)体系。J2SE就是Java2的标准版,主要用于桌面应用软件的编程;J2ME主要应用于嵌入是系统开发,如手机和PDA的编程;J2EE
- APP推广
aoyouzi
APP推广
一,免费篇
1,APP推荐类网站自主推荐
最美应用、酷安网、DEMO8、木蚂蚁发现频道等,如果产品独特新颖,还能获取最美应用的评测推荐。PS:推荐简单。只要产品有趣好玩,用户会自主分享传播。例如足迹APP在最美应用推荐一次,几天用户暴增将服务器击垮。
2,各大应用商店首发合作
老实盯着排期,多给应用市场官方负责人献殷勤。
3,论坛贴吧推广
百度知道,百度贴吧,猫扑论坛,天涯社区,豆瓣(
- JSP转发与重定向
百合不是茶
jspservletJava Webjsp转发
在servlet和jsp中我们经常需要请求,这时就需要用到转发和重定向;
转发包括;forward和include
例子;forwrad转发; 将请求装法给reg.html页面
关键代码;
req.getRequestDispatcher("reg.html
- web.xml之jsp-config
bijian1013
javaweb.xmlservletjsp-config
1.作用:主要用于设定JSP页面的相关配置。
2.常见定义:
<jsp-config>
<taglib>
<taglib-uri>URI(定义TLD文件的URI,JSP页面的tablib命令可以经由此URI获取到TLD文件)</tablib-uri>
<taglib-location>
TLD文件所在的位置
- JSF2.2 ViewScoped Using CDI
sunjing
CDIJSF 2.2ViewScoped
JSF 2.0 introduced annotation @ViewScoped; A bean annotated with this scope maintained its state as long as the user stays on the same view(reloads or navigation - no intervening views). One problem w
- 【分布式数据一致性二】Zookeeper数据读写一致性
bit1129
zookeeper
很多文档说Zookeeper是强一致性保证,事实不然。关于一致性模型请参考http://bit1129.iteye.com/blog/2155336
Zookeeper的数据同步协议
Zookeeper采用称为Quorum Based Protocol的数据同步协议。假如Zookeeper集群有N台Zookeeper服务器(N通常取奇数,3台能够满足数据可靠性同时
- Java开发笔记
白糖_
java开发
1、Map<key,value>的remove方法只能识别相同类型的key值
Map<Integer,String> map = new HashMap<Integer,String>();
map.put(1,"a");
map.put(2,"b");
map.put(3,"c"
- 图片黑色阴影
bozch
图片
.event{ padding:0; width:460px; min-width: 460px; border:0px solid #e4e4e4; height: 350px; min-heig
- 编程之美-饮料供货-动态规划
bylijinnan
动态规划
import java.util.Arrays;
import java.util.Random;
public class BeverageSupply {
/**
* 编程之美 饮料供货
* 设Opt(V’,i)表示从i到n-1种饮料中,总容量为V’的方案中,满意度之和的最大值。
* 那么递归式就应该是:Opt(V’,i)=max{ k * Hi+Op
- ajax大参数(大数据)提交性能分析
chenbowen00
WebAjax框架浏览器prototype
近期在项目中发现如下一个问题
项目中有个提交现场事件的功能,该功能主要是在web客户端保存现场数据(主要有截屏,终端日志等信息)然后提交到服务器上方便我们分析定位问题。客户在使用该功能的过程中反应点击提交后反应很慢,大概要等10到20秒的时间浏览器才能操作,期间页面不响应事件。
根据客户描述分析了下的代码流程,很简单,主要通过OCX控件截屏,在将前端的日志等文件使用OCX控件打包,在将之转换为
- [宇宙与天文]在太空采矿,在太空建造
comsci
我们在太空进行工业活动...但是不太可能把太空工业产品又运回到地面上进行加工,而一般是在哪里开采,就在哪里加工,太空的微重力环境,可能会使我们的工业产品的制造尺度非常巨大....
地球上制造的最大工业机器是超级油轮和航空母舰,再大些就会遇到困难了,但是在空间船坞中,制造的最大工业机器,可能就没
- ORACLE中CONSTRAINT的四对属性
daizj
oracleCONSTRAINT
ORACLE中CONSTRAINT的四对属性
summary:在data migrate时,某些表的约束总是困扰着我们,让我们的migratet举步维艰,如何利用约束本身的属性来处理这些问题呢?本文详细介绍了约束的四对属性: Deferrable/not deferrable, Deferred/immediate, enalbe/disable, validate/novalidate,以及如
- Gradle入门教程
dengkane
gradle
一、寻找gradle的历程
一开始的时候,我们只有一个工程,所有要用到的jar包都放到工程目录下面,时间长了,工程越来越大,使用到的jar包也越来越多,难以理解jar之间的依赖关系。再后来我们把旧的工程拆分到不同的工程里,靠ide来管理工程之间的依赖关系,各工程下的jar包依赖是杂乱的。一段时间后,我们发现用ide来管理项程很不方便,比如不方便脱离ide自动构建,于是我们写自己的ant脚本。再后
- C语言简单循环示例
dcj3sjt126com
c
# include <stdio.h>
int main(void)
{
int i;
int count = 0;
int sum = 0;
float avg;
for (i=1; i<=100; i++)
{
if (i%2==0)
{
count++;
sum += i;
}
}
avg
- presentModalViewController 的动画效果
dcj3sjt126com
controller
系统自带(四种效果):
presentModalViewController模态的动画效果设置:
[cpp]
view plain
copy
UIViewController *detailViewController = [[UIViewController al
- java 二分查找
shuizhaosi888
二分查找java二分查找
需求:在排好顺序的一串数字中,找到数字T
一般解法:从左到右扫描数据,其运行花费线性时间O(N)。然而这个算法并没有用到该表已经排序的事实。
/**
*
* @param array
* 顺序数组
* @param t
* 要查找对象
* @return
*/
public stati
- Spring Security(07)——缓存UserDetails
234390216
ehcache缓存Spring Security
Spring Security提供了一个实现了可以缓存UserDetails的UserDetailsService实现类,CachingUserDetailsService。该类的构造接收一个用于真正加载UserDetails的UserDetailsService实现类。当需要加载UserDetails时,其首先会从缓存中获取,如果缓存中没
- Dozer 深层次复制
jayluns
VOmavenpo
最近在做项目上遇到了一些小问题,因为架构在做设计的时候web前段展示用到了vo层,而在后台进行与数据库层操作的时候用到的是Po层。这样在业务层返回vo到控制层,每一次都需要从po-->转化到vo层,用到BeanUtils.copyProperties(source, target)只能复制简单的属性,因为实体类都配置了hibernate那些关联关系,所以它满足不了现在的需求,但后发现还有个很
- CSS规范整理(摘自懒人图库)
a409435341
htmlUIcss浏览器
刚没事闲着在网上瞎逛,找了一篇CSS规范整理,粗略看了一下后还蛮有一定的道理,并自问是否有这样的规范,这也是初入前端开发的人一个很好的规范吧。
一、文件规范
1、文件均归档至约定的目录中。
具体要求通过豆瓣的CSS规范进行讲解:
所有的CSS分为两大类:通用类和业务类。通用的CSS文件,放在如下目录中:
基本样式库 /css/core
- C++动态链接库创建与使用
你不认识的休道人
C++dll
一、创建动态链接库
1.新建工程test中选择”MFC [dll]”dll类型选择第二项"Regular DLL With MFC shared linked",完成
2.在test.h中添加
extern “C” 返回类型 _declspec(dllexport)函数名(参数列表);
3.在test.cpp中最后写
extern “C” 返回类型 _decls
- Android代码混淆之ProGuard
rensanning
ProGuard
Android应用的Java代码,通过反编译apk文件(dex2jar、apktool)很容易得到源代码,所以在release版本的apk中一定要混淆一下一些关键的Java源码。
ProGuard是一个开源的Java代码混淆器(obfuscation)。ADT r8开始它被默认集成到了Android SDK中。
官网:
http://proguard.sourceforge.net/
- 程序员在编程中遇到的奇葩弱智问题
tomcat_oracle
jquery编程ide
现在收集一下:
排名不分先后,按照发言顺序来的。
1、Jquery插件一个通用函数一直报错,尤其是很明显是存在的函数,很有可能就是你没有引入jquery。。。或者版本不对
2、调试半天没变化:不在同一个文件中调试。这个很可怕,我们很多时候会备份好几个项目,改完发现改错了。有个群友说的好: 在汤匙
- 解决maven-dependency-plugin (goals "copy-dependencies","unpack") is not supported
xp9802
dependency
解决办法:在plugins之前添加如下pluginManagement,二者前后顺序如下:
[html]
view plain
copy
<build>
<pluginManagement