开篇致谢:下面内容的整个框架是跟着生信技能树发布在bilibili上的ChIP-seq视频学习的,链接:https://www.bilibili.com/video/av29255401,如果有基础的话,应该可以很快学下来。
另外,用到的测试数据来自我之前介绍的一篇文献,拟南芥的,跑起来快。链接:https://www.jianshu.com/p/24dbcbcd26e5,后面关于ChIP-seq在组蛋白修饰中应用的学习就以这一篇为主。另外,在转录因子中的应用以及ATAC-seq我在这个暑假都会学一遍。
1. 数据获取
###下载SRA数据
for sample in SRR519051{3..8}
do
prefetch $sample
done
###解压,并更改样本名称
$ cat config
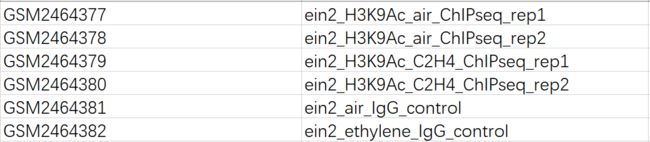
ein2-5_air_H3K9aC_1 SRR5190513
ein2-5_air_H3K9aC_2 SRR5190514
ein2-5_ethylene_H3K9aC_1 SRR5190515
ein2-5_ethylene_H3K9aC_2 SRR5190516
ein2-5_air_MPGB_1 SRR5190517
ein2-5_ethylene_MPGB_1 SRR5190518
dir=~/ChIP_seq/raw
cat config | while read id
do
echo $id #如何将变量赋值给变量?
array=($id)
srr=${array[1]}
sample_name=${array[0]}
fastq-dump -A $sample_name -O $dir --gzip --split-3 ~/ChIP_seq/sra/$srr.sra
done
###查看
$ ls
ein2-5_air_H3K9aC_1.fastq.gz ein2-5_air_H3K9aC_2.fastq.gz ein2-5_air_MPGB_1.fastq.gz ein2-5_ethylene_H3K9aC_1.fastq.gz ein2-5_ethylene_H3K9aC_2.fastq.gz ein2-5_ethylene_MPGB_1.fastq.gz
huangsiyuan 13:17:41 ~/ChIP_seq/raw
2. 质量检测
ls ~/ChIP_seq/raw/*.fastq.gz | xargs fastqc -t 4
multiqc ~/ChIP_seq/raw/
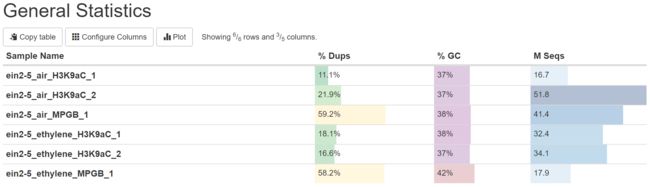
最终的结果表明这6个数据的质量都很好,所以原文并没有进行额外的过滤操作。需要注意的是,在该文献中ein2-5_air_MPGB_1和ein2-5_ethylene_MPGB_1两个阴性对照呈现出reads重复率高的特征
3. 比对
3.1 参考基因组建立索引
gunzip Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
bowtie-build Arabidopsis_thaliana.TAIR10.dna.toplevel.fa Arabidopsis_thaliana.TAIR10.dna.toplevel
3.2 比对
bowtie_index=/ifs1/Grp3/huangsiyuan/Ref/Arabidopsis/Arabidopsis_thaliana.TAIR10.dna.toplevel
cd ~/ChIP_seq/align/
ls ~/ChIP_seq/raw/*.fastq.gz | while read id
do
file=$(basename $id)
bam_name=${file%%.*}
bowtie -p 4 -M 1 --best --strata $bowtie_index $id -S | samtools sort -O bam -@ 4 - > ${bam_name}.bam
done
-M 1 --best --strata的作用是将多比对中得分最高的一条比对记录保留,原文在这里采用的是保留唯一比对的reads记录,有些区别。
3.3 比对率查看
针对
-M 1 --best --strata这一种模式。允许多比对的话,下面的等式关系不一定成立。
bowtie比对完之后会在log文件中报告比对率,比如
# reads processed: 16679255
# reads with at least one reported alignment: 15164883 (90.92%) #比对得上,不管是唯一比对还是多比对,(都只会报告一条比对记录)
# reads that failed to align: 250108 (1.50%) #完全比对不上
# reads with alignments sampled due to -M: 1264264 (7.58%) #这1264264是指哪一些reads,自己的理解是:这些reads多比对,但不能按照优劣排序(比如有两个以上最优),故不予考虑。之前做小RNA有类似的处理,当reads多比对次数超过一个阈值,就不考虑这个reads了。
Reported 15164883 alignments
还可以用samtools flagstat简单统计比对率
16679255 + 0 in total (QC-passed reads + QC-failed reads)
16429147 + 0 mapped (98.50% : N/A) #这个值是上面的90.92%和7.58%的和。
又统计了一下fq里面的reads数量
$ samtools view ein2-5_air_H3K9aC_1.bam | wc -l
16679255 #bam文件有多少条比对记录
$ samtools view ein2-5_air_H3K9aC_1.bam | cut -f1 | sort | uniq | wc -l
16679255 #这些记录对应多少条reads,相等说明:一条reads只对应一条记录
$ lsx ein2-5_air_H3K9aC_1.fastq.gz | wc -l
66717020
$ echo "scale=2; 66717020/4" | bc
16679255.00 #相等说明:不管reads有没有比对上,都会在bam中有一个记录
这一步没有什么实质性的作用,但能加深对reads比对率、唯一比对、多比对、reads数、比对记录数等的理解。
4. 去除PCR重复
"Duplicate reads were removed using SAMtools."相关的NC文章是这样做的。
ls *bam | while read id; do samtools markdup -r $id $(basename $id ".bam").sm.bam & done
5. 合并bam
按照文章说的,相同处理下,探究相同的组蛋白修饰,将不同的重复合并再进行后面的处理
samtools merge ein2-5_ethylene_H3K9aC_merge.sm.bam ein2-5_ethylene_H3K9aC_1.sm.bam ein2-5_ethylene_H3K9aC_2.sm.bam
samtools merge ein2-5_air_H3K9aC_merge.sm.bam ein2-5_air_H3K9aC_1.sm.bam ein2-5_air_H3K9aC_2.sm.bam
并用samtools index为4个待分析的bam建立索引。
6. MACS2找peaks
文章只强调了--nomodel, -p 0.01这两个参数。
macs2 callpeak --nomodel -B -p 0.01 -c ein2-5_air_MPGB_1.sm.bam -t ein2-5_air_H3K9aC_merge.sm.bam -f BAM -g 110000000 -n ein2-5_air_H3K9aC
macs2 callpeak --nomodel -B -p 0.01 -c ein2-5_ethylene_MPGB_1.sm.bam -t ein2-5_ethylene_H3K9aC_merge.sm.bam -f BAM -g 110000000 -n ein2-5_ethylene_H3K9aC
$ ll ein2-5_*
-rw-r--r--. 1 huangsiyuan grp3 332M Jul 9 13:49 ein2-5_air_H3K9aC_control_lambda.bdg #control的可视化文件
-rw-r--r--. 1 huangsiyuan grp3 1.5M Jul 9 13:49 ein2-5_air_H3K9aC_peaks.narrowPeak #peaks信息
-rw-r--r--. 1 huangsiyuan grp3 1.7M Jul 9 13:49 ein2-5_air_H3K9aC_peaks.xls #peaks信息
-rw-r--r--. 1 huangsiyuan grp3 1.1M Jul 9 13:49 ein2-5_air_H3K9aC_summits.bed #summits(峰顶)的位置
-rw-r--r--. 1 huangsiyuan grp3 1.4G Jul 9 13:49 ein2-5_air_H3K9aC_treat_pileup.bdg #处理的可视化文件
-rw-r--r--. 1 huangsiyuan grp3 149M Jul 9 13:58 ein2-5_ethylene_H3K9aC_control_lambda.bdg
-rw-r--r--. 1 huangsiyuan grp3 1.3M Jul 9 13:58 ein2-5_ethylene_H3K9aC_peaks.narrowPeak
-rw-r--r--. 1 huangsiyuan grp3 1.4M Jul 9 13:58 ein2-5_ethylene_H3K9aC_peaks.xls
-rw-r--r--. 1 huangsiyuan grp3 910K Jul 9 13:58 ein2-5_ethylene_H3K9aC_summits.bed
-rw-r--r--. 1 huangsiyuan grp3 1.3G Jul 9 13:58 ein2-5_ethylene_H3K9aC_treat_pileup.bdg
7. 可视化
7.1 deeptools
先将bam->bw
ls *sm.bam | while read id; do bamCoverage --binSize 50 --normalizeUsing RPKM -b $id -o ${id%_*}.bw done
看参数的值就能知道bw存储的是什么信息:横坐标是在基因组上的一对起始位置,窗口大小是50bp,纵坐标是将深度标准化之后得到的RPKM。
除了bamCoverage,bamCompare也能将bam->bw,并且同时考虑处理和对照,以消除噪声。原文是这样说的:To show ChIP binding signal surrounding TSSs or in gene bodies, read coverage was first calculated using the bamCompare tool (RPKM, Log2(ChIP/IgG) in deepTools.
bamCompare -b1 ein2-5_air_H3K9aC_merge.sm.bam -b2 ein2-5_air_MPGB_1.sm.bam \
--operation log2 --normalizeUsing RPKM --scaleFactorsMethod None --binSize 50 \
-o ein2-5_air_H3K9aC_MPGB_log2ratio.bw
bamCompare -b1 ein2-5_ethylene_H3K9aC_merge.sm.bam -b2 ein2-5_ethylene_MPGB_1.sm.bam \
--operation log2 --normalizeUsing RPKM --scaleFactorsMethod None --binSize 50 \
-o ein2-5_ethylene_H3K9aC_MPGB_log2ratio.bw
查看TSS位点附近的信号
#TSS位点如何确认?five_prime_UTR的第一个位置?那如果一个基因有多个转录本多个five_prime_UTR怎么办?每个都看?
ls *bw | while read id
do
computeMatrix reference-point --referencePoint TSS -a 3000 -b 3000 -R ~/Ref/Arabidopsis/Arabidopsis_thaliana.TAIR10.44.gtf -S $id --skipZeros -o ${id%%.*}.matrix.TSS.3k.gz --outFileSortedRegions ${id%%.*}.region.TSS.3k.bed -p 4
plotHeatmap -m ${id%%.*}.matrix.TSS.3k.gz -o ${id%%.*}.png
plotHeatmap -m ${id%%.*}.matrix.TSS.3k.gz -o ${id%%.*}.pdf --plotFileFormat pdf --dpi 720
plotProfile -m ${id%%.*}.matrix.TSS.3k.gz -o ${id%%.*}_2.png
plotProfile -m ${id%%.*}.matrix.TSS.3k.gz -o ${id%%.*}_2.pdf --plotFileFormat pdf --perGroup --dpi 720
done
查看gene_body的信号
ls ~/ChIP_seq/align/*bw | while read id
do
computeMatrix scale-regions -a 3000 -b 3000 --regionBodyLength 6000 -R ~/Ref/Arabidopsis/Arabidopsis_thaliana.TAIR10.44.gtf -S $id --skipZeros -o $(basename $id ".bw").matrix.TSS_TES.3k.gz --outFileSortedRegions $(basename $id ".bw").region.TSS_TES.3k.bed -p 4
plotHeatmap -m $(basename $id ".bw").matrix.TSS_TES.3k.gz -o $(basename $id ".bw").png
plotHeatmap -m $(basename $id ".bw").matrix.TSS_TES.3k.gz -o $(basename $id ".bw").pdf --plotFileFormat pdf --dpi 720
plotProfile -m $(basename $id ".bw").matrix.TSS_TES.3k.gz -o $(basename $id ".bw")_2.png
plotProfile -m $(basename $id ".bw").matrix.TSS_TES.3k.gz -o $(basename $id ".bw")_2.pdf --plotFileFormat pdf --perGroup --dpi 720
done

7.2 IGV
在igv下面看看bed, bam, bw, bgd是什么样子
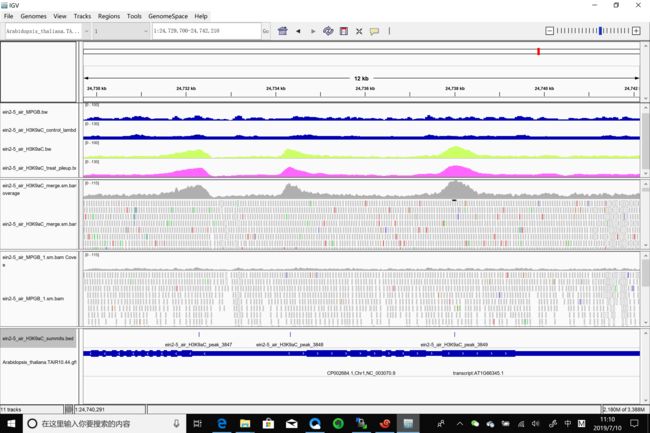
可以看到,bam, bw, bdg整体趋势一致,并且跟对照相比,可以显示出peaks。
8. peaks注释
针对的是summits.bed文件,看peaks(一个峰,精确到了单碱基位置)属于哪一个基因?或是离哪一个基因最近?属于哪一个区域?
用chipseeker, 在R下面操作
library(ChIPseeker)
require(GenomicFeatures)
ath <- makeTxDbFromGFF("Arabidopsis_thaliana.TAIR10.44.gff3")
bedpeaksfile = "ein2-5_air_H3K9aC_summits.bed"
peak <- readPeakFile(bedpeaksfile)
peakAnno <- annotatePeak(peak, tssRegion = c(-3000, 3000), TxDb = ath)
plotAnnoPie(peakAnno)
plotAnnoBar(peakAnno)
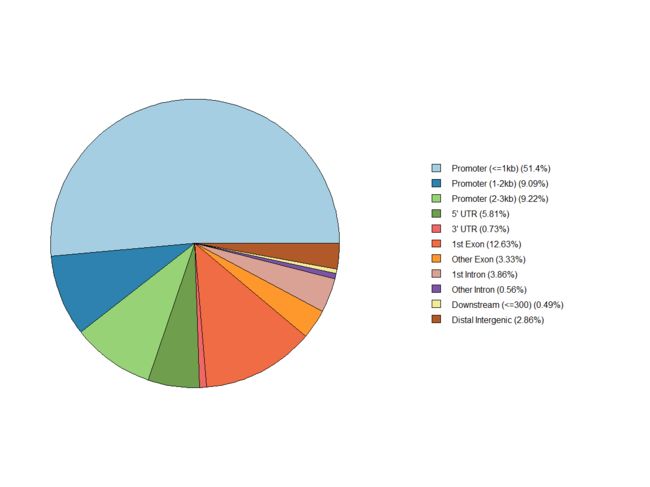

这个summits的注释跟变异检测注释原理差不多,只是分区不一样。
寻找motif相关的内容,这篇组蛋白修饰的文献没有涉及到,后面再做一个转录因子的再学学。