Chip-seq流程报告
一、摘要
实验旨在了解Chip-seq的基本原理。通过模仿文献《Targeting super enhancer associated oncogenes in oesophageal squamous cell carcinoma》的流程,学会利用NCBI和EBI数据库下载数据,熟悉Linux下的基本操作,并使用R语言画图,用Python或者shell写脚本进行基本的数据处理,通过FastQC、Bowtie、Macs、samtools、ROSE等软件进行数据处理,并对预测结果进行分析讨论。
二、材料
1、硬件平台
处理器:Intel(R) Core(TM)i7-4710MQ CPU @ 2.50GHz 2.50GHz
安装内存(RAM):16.0GB
2、系统平台
Windows 8.1,Ubuntu
3、软件平台
① Aspera connect ② FastQC ③ Bowtie
④ Macs 1.4.2 ⑤ IGV ⑥ ROSE
4、数据库资源
NCBI数据库:https://www.ncbi.nlm.nih.gov/;
EBI数据库:http://www.ebi.ac.uk/;
5、研究对象
加入H3K27Ac 抗体处理过的TE7细胞系测序数据和其空白对照组
加入H3K27Ac 抗体处理过的KYSE510细胞系和其空白对照组
背景简介:食管鳞状细胞癌(OSCC)是一种侵袭性的恶性肿瘤,本文章通过高通量小分子抑制剂进行筛选,发现了一个高度有效的抗癌物,特异性的CDK7抑制剂THZ1。RNA-Seq显示,低剂量THZ1会对一些致癌基因的产生选择性抑制作用,而且,对这些THZ1敏感的基因组功能的进一步表征表明他们经常与超级增强子结合(SE)。ChIP-seq解读在OSCC细胞中,CDK7的抑制作用的机制。
本文亮点:确定了在OSCC细胞中SE的位置,以及识别出许多SE有关的调节元件;并且发现小分子THZ1特异性抑制SE有关的转录,显示强大的抗癌性。
文章PMID: 27196599
三、方法
1、Aspera软件下载及安装
进入Aspera官网的Downloads界面,选中aspera connect server,点击Wwindows图标,选择v3.6.2版本,点击Download进行下载。
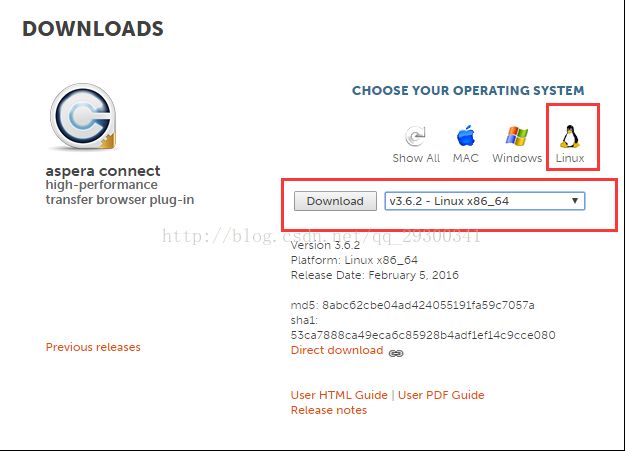
图表 1 aspera的下载
Linux下的安装配置参考博文:
http://blog.csdn.net/likelet/article/details/8226368
2、Chip-Seq数据下载
1)选择NCBI的GEO DataSets数据库,输入GSE76861,打开GSM2039110、GSM2039111、2039112、GSM2039113获取它们对应的SRX序列号。

图表 2 Chip-seq数据

图表 3 获取SRA编号
2)进入EBI,获取ascp下载地址
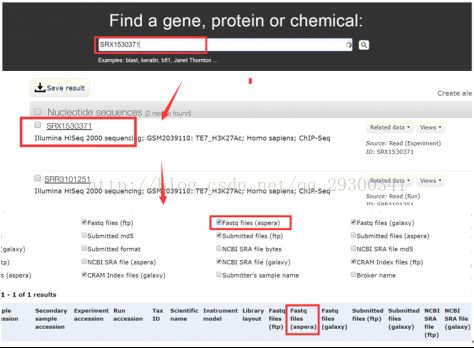
图表 4 ascp下载地址
3)使用aspera下载并解压
aspera下载命令及gunzip解压命令(nohup+命令+&可以后台运行)

3、FastQC质量检查
3.1 FastQC的安装
Ubuntu软件包内自带Fastqc
故安装命令apt-get install fastqc
3.2 使用FastQC进行质量检查
fastqc命令:
fastqc -o . -t 5 -f fastq SRR3101251.fastq &
-o . 将结果输出到当前目录
-t 5 表示开5个线程运行
-f fastq SRR3101251.fastq 表示输入的文件
(要分别对四个fastq文件执行四次)
4、使用Bowtie对Reads进行Mapping
4.1 Bowtie的安装
Ubuntu软件包内自带bowtie
故安装命令apt-get install bowtie
4.2 下载人类参考基因组
文献说序列比对到了人类参考基因组GRCh37/hg19上
bowtie官网上面有人类参考基因组hg19已经建好索引的文件
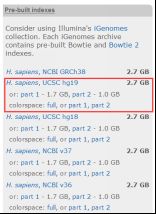
图表 5 bowtie hg19建好的索引
再执行解压缩命令:unzip hg19.ebwt.zip
4.3 使用bowtie进行比对
bowtie命令:

5、MACS寻找Peak富集区
5.1 Macs14的安装
至刘小乐实验室网站下载http://liulab.dfci.harvard.edu/MACS/Download.html

解压后,切换到文件夹目录,执行
python setup.py install
5.2 使用Macs建模,寻找Peaks富集区
MACS命令:

6、IGV可视化
6.1数据正规化normalised
编写python程序对wig文件进行normalised

对TE7_H3K27Ac和KYSE510_H3K27Ac的wig文件(即MACS后生成的treat文件夹里的wig文件)计算RPM
RPM公式:(某位置的reads数目÷所有染色体上总reads数目)×1000000
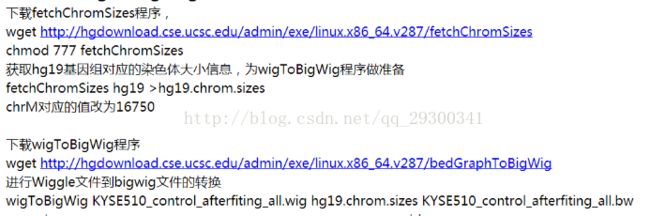
6.2 使用wigToBigWig转化格式
6.3安装IGV(Integrative Genomics Viewer)对结果可视化
从IGV官网下载windows版本http://software.broadinstitute.org/software/igv/download根据提示安装
直接点击打开igv.jar或者对bat文件以管理员身份运行
首先,载入hg19基因组;接着载入两个normalised后的bw文件即可
7、ROSE鉴定Enhancer
7.1 ROSE程序安装
ROSE程序可以到http://younglab.wi.mit.edu/super_enhancer_code.html下载,并且有2.7G的示例数据
7.2 数据预处理

7.3运行ROSE程序

7.4 进行基因注释
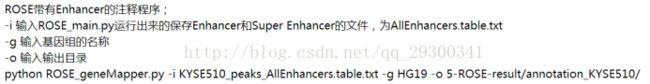
7.5 编写R程序,绘制Enhancer及邻近基因

图表 6 TE7.r程序

图表 7 KYSE510.r程序
四、结果
1、Chip-Seq数据下载
Chip-Seq数据下载并解压结果
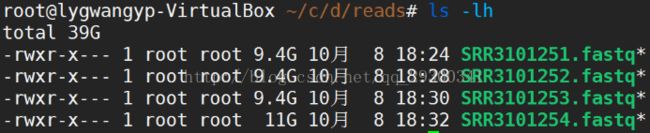
图表 8 Chip-Seq数据
2、FastQC质量检查
数据质量检查

图表 9 质量检查文件

图表 10 质量检查结果
3、使用Bowtie对Reads进行Mapping
3.1基因组文件
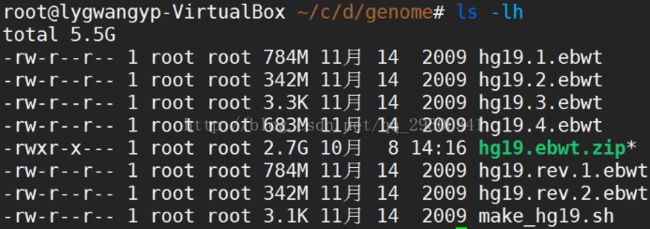
图表 11人类参考基因组HG19索引
3.2 Mapping结果

图表 12 Mapping整体结果
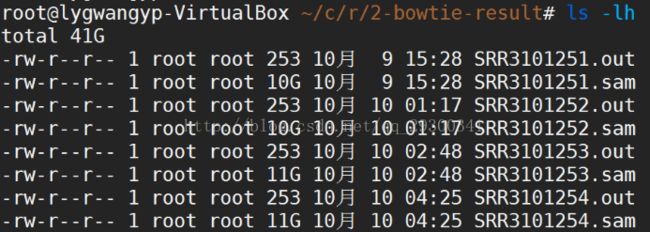
图表 13 生成的sam文件
4、MACS寻找Peak富集区
4.1MACS结果文件

图表 14 TE7实验对照组结果

图表 15 KYSE510实验对照组结果
4.2 MACS结果解读
Peaks.xls从左至右依次是:峰所在的染色体名称,峰的起始位置,峰的结束为止,峰的长度,峰的高度,贴上的reads标签个数,pvalue(表示置信度),峰的富集程度,FDR假阳性率(越小则峰越好)

图表 16 Peaks.xls文件
negative_peaks.xls当有对照组实验存在时,MACS会进行两次peak calling。第一次以实验组(Treatment)为实验组,对照组为对照组,第二次颠倒,以实验组为对照组,对照组为实验组。这个相当于颠倒过后计算出来的文件

图表 17 negative_peaks.xls
Peaks.bed文件相当于Peaks.xls的简化版,从左至右依次是:峰所在的染色体名称,峰的起始位置,峰的结束为止,峰的MACS名称,pvalue(表示置信度)
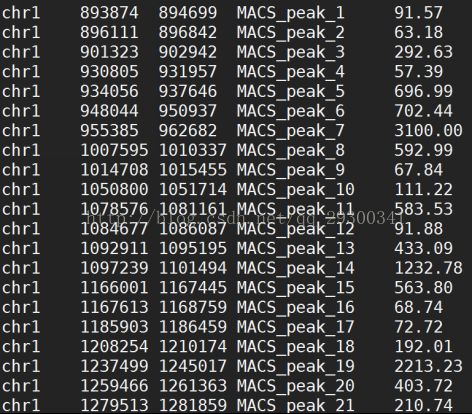
图表 18 Peaks.bed文件
summits.bed是峰顶文件,从左至右依次是:峰所在的染色体名称,峰顶的位置,峰的MACS名称,峰的高度
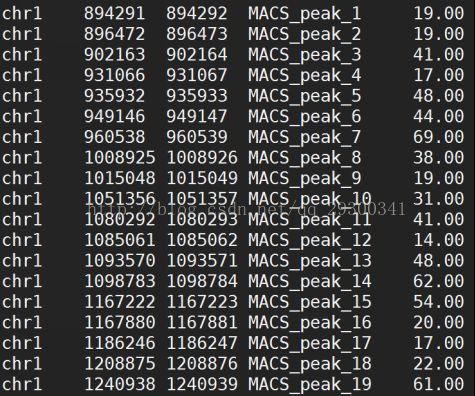
图表 19 summits.bed文件
MACS_wiggle文件夹下面分为control文件夹和treat文件夹,里面分别存了control组和treat组每隔50bp,贴上的reads数目。第一列为染色体上的位置;第二列为从第一列对应的位置开始,延伸50bp,总共贴上的标签(reads)个数。

图表 20 wiggle文件夹下afterfiting_all.wig文件
model.r文件可以使用R运行,绘制双峰模型的图片PDF
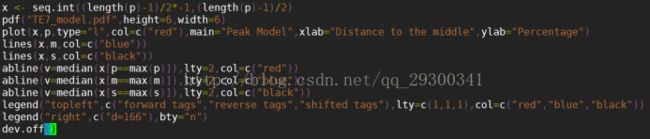
图表 21 model.r文件


图表 22 TE7双峰模型 图表 23 KYSE510双峰模型
5、IGV对peaks可视化
5.1Normalised后,wig文件与文献数据比较

图表 24 peaks整体统计比较
5.2 IGV peaks整体可视化

图表 25 IGV可视化
6、ROSE分析结果
6.1 数据预处理结果
Samtools将sam文件转化为bam文件,并且排序,再建立索引

图表 26 bam文件和bai索引
6.2 ROSE程序Enhancer分类结果

图表 27 TE7 Enhancer分类结果
图表 28 KYSE510 Enhancer分类结果
peaks_AllEnhancers.table.txt文件从左到右分别是,Enhancer区域名称ID,染色体位置,Enhancer起始位置,结束位置,由多少个Enhancer缝合连接而成,Enhancer大小,Treat组峰高度,Control组峰高度,Enhancer大小排名,是否为Super Enhancer

图表 29 peaks_AllEnhancers.table.txt文件
peaks_Plot_points.png图片,纵坐标为peaks_AllEnhancers.table.txt中G,H列相减结果,及减掉对照组峰后的高度,横坐标为全部Enhancer的排名,越可能是SuperEnhancer则越靠图的右边。

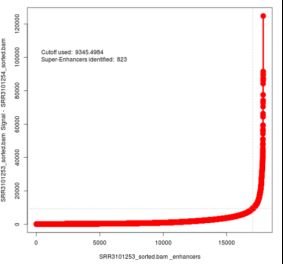
图表 30 TE7_peaks_Plot_points.png图表 31 KYSE510_peaks_Plot_points.png
6.3 基因注释结果
AllEnhancers_ENHANCER_TO_GENE.txt第J列开始为离Enhancer最近的基因名称
AllEnhancers_GENE_TO_ENHANCER.txt第1列为基因名,后面为邻近峰的名称
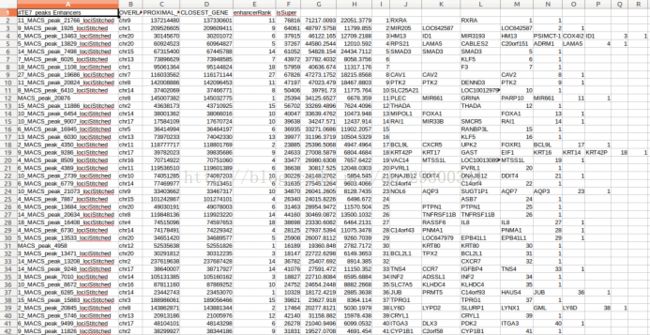
图表 32 AllEnhancers_ENHANCER_TO_GENE.txt文件
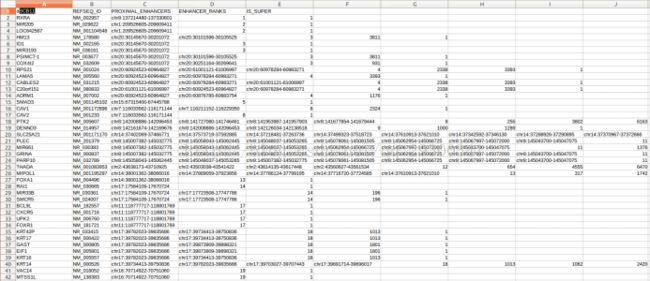
图表 33 AllEnhancers_GENE_TO_ENHANCER.txt
五、讨论和结论
1、结论
1.1 FastQC质量检查
FastQC 版本和机房小型机不同,为v0.10.1,因此检测结果略有区别。图表 8 质量检查结果显示,测序质量挺好,Per base sequence content、Per sequence GC content、Kmer Content出现警告更可能是由于测序方法本身存在的固有误差。
1.2 bowtie整体覆盖度
由图表 10 Mapping整体结果可以看出,四个fastq文件Mapping整体覆盖率都在90%以上,从另一方面说明数据质量很好
1.3 ROSE辨别出的Super Enhancer
由图表 29 TE7_peaks_Plot_points.png图表 28 KYSE510_peaks_Plot_points.png可以看出,在TE7细胞系中,找出了439个Super Enhancer,在KYSE510细胞系中,找出了823个Super Enhancer。
2、讨论
由IGV可视化图可以看出,峰的高度和位置基本和文献相同。
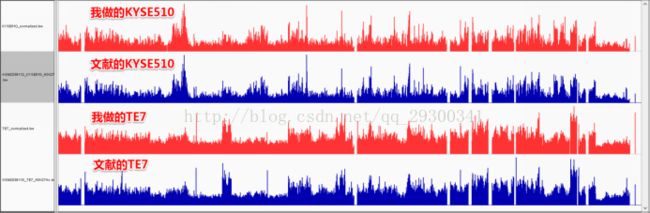
图表 34 IGV可视化图
再用R程序根据ROSE程序结果,绘制和文献相同的图片,与文献的图片进行比较,可以看出来,基因的分布是相似的,就是具体位置和文献不是很一样。
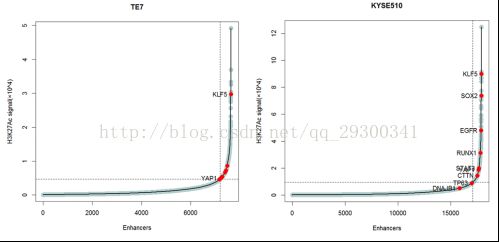
图表 35 本流程结果

图表 36 文献结果
在MACS结果中,有些很窄的峰高度明显比文献要低,这可能是因为bowtie时候,设置的参数使得多条reads比对上仅输出一次,使得峰高度减小。
在ROSE结果中,MIR205HG没有标注出来,而文献中有此基因,经过检查,在相似位置ROSE程序有找到MIR205基因,这可能是基因注释文件和文献不同导致的。
参考文献
[1] Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma PMID: 27196599