中文题目:微生物组研究中优化方法与规避误区
发表期刊:Microbiome
发表时间:2017年
影响因子:10.465
摘要:
本文提出了微生物组优化实验设计和规避误区的建议,综述了实验设计的最佳方法;介绍了动物研究中常见的影响因素,对几种样本类型提供了最佳储存方法;
讨论了应结合实验样本的阳性和阴性对照进行设计和分析;介绍了可用作阳性对照的非生物DNA序列;
低微生物量的样品研究中,仔细分析阴性和阳性对照尤其重要,因为污染可能导致部分或全部样本组成发生改变;
总结了通过严格多重比较控制以及比较发现队列和验证队列来提高实验稳健性的方法。
研究背景
近年来,微生物群落—微生物组的研究变得非常流行。通过高通量测序方法,我们可以获得不同来源的微生物群落的组成,并通过比较来揭示微生物群的相关模式。在实验和分析中,所有的分析方法都有可能产生偏差。例如,16S rRNA基因片段分析时,基因区域的选择会影响细菌类型。如果样本中微生物DNA量很少,文库准备和测序方法通常会得到来自污染的序列。污染序列可能来源于试剂、灰尘、样品间或其他来源的交叉污染。
本综述旨在帮助微生物组研究人员解决这些问题,我们主要集中在微生物组分析实验室工作上,并只简要介绍计算和统计方法。我们按照实验顺序来呈现各部分内容:实验设计,样本收集处理和数据分析。
一、微生物组实验设计
我们必须认真设计实验以确保所进行的实验能够回答所提出的问题。从一开始就要设计研究所涉及的统计分析方法。如果可能的话,可以进行功效分析。
影响因素
· 抗生素使用
抗生素治疗会对肠道菌群产生影响[5,40-42];使用质子泵抑制剂会改变下消化道微生物菌群的组成,增加艰难梭菌感染的风险。
· 饮食
微生物群落结构和基因表达会在短期内发生变化,以响应饮食中的极端短期变化[57]。长期的饮食模式与某些特定菌属主导的肠道菌群有关:高蛋白和动物脂肪的饮食与高丰度拟杆菌属相关,而高碳水化合物的饮食与高丰度的普雷沃氏菌属有关[55]。
· 年龄
通常情况下,人类3岁左右的肠道菌群采用稳定的厌氧模式,但在早期生命中有所不同[58-60]。老年人通常会拥有高丰度的变形菌门[61]。因此,使用年龄匹配的对照进行微生物菌群比较研究是至关重要的。
· 宠物饲养
研究表明,狗主人和狗会具有相似的皮肤菌群[69]。
· 性别
早期接触微生物能够提高雄性小鼠体内的睾酮水平,对1型糖尿病具有保护作用。将受保护的雄性小鼠的微生物菌群移植到年轻雌性小鼠时,对1型糖尿病的保护作用是相同的[63]。小鼠体内的微生物昼夜节律在不同性别之间存在差异[65]。值得注意的是,男性的性偏好也与肠道菌群的差异有关[67],这可能是肠道菌群和HIV感染研究中的一个混杂因素。
每一个因素如何影响给定的微生物组研究主要取决于提出的问题和研究组间差异程度。在实验设计期间列举可能的影响因素并进行量化,统计分析中将它们作为独立变量处理。
纵向不稳定性(时间梯度)
随着时间的推移,健康成人的肠道菌群组成基本上是稳定的[70-72],而菌群失调与炎症性肠道疾病等疾病有关[1,5,73]。肠道菌群在每天也表现出昼夜节律行为[65,79,80]。然而人体其他部位,如阴道菌群可以在短期内发生极大变化[74-78]。因此,研究一种新的样本类型,了解它的菌群纵向变化以筛选能解决问题的样本是至关重要的。
不同批次的DNA提取试剂盒可能是纵向研究的重要变异来源[23,81]。明智的做法是在研究开始时购买所有需要的提取试剂盒,或者同时存储样品和提取所有样品,从而将此变量的影响降至最低。
动物实验的笼子效应
笼子效应会影响小鼠的微生物菌群研究,对其他动物也同样重要。同一笼子里的小鼠由于食粪性而共享相似的肠道菌群[82]。研究发现老鼠品系可以解释肠道菌群19%的变异,而笼子效应可以解释31%[83]。研究人员必须为每个研究组设置多个笼子,并在统计分析中将笼子作为一个变量。根据笼子变量的检测效果来确定不同组间的微生物群落是否存在差异。为了降低成本,每笼可以饲养两到三只老鼠[84-86]。
小鼠抗生素干预期间真菌种群的纵向研究发现,抗生素治疗与治疗组中真菌定植的增加有关(图1)。检测到的真菌在每个笼子内基本一致,但在每个处理组和未处理对照组中,各笼子之间的真菌检测结果也有所不同。检测到的真菌类型在纵向上有所变化,但在笼内是一致的。

二、样品采集和处理过程中注意事项
样品储存条件
微生物组样本保存过程中最重要的是减少从样本采集到处理过程中原始微生物菌群的变化,且所有样本的保存条件保持一致。
通过比较−80°C下立即冷冻、冰上保存24小时或冰上保存48小时后提取和分析的人类粪便样本,发现与不同个体间的差异相比,储存方法的差异并不显著[88]。

我们收集了三名健康受试者的口腔拭子样本,并将其保存在不同条件下(在−20°C冷冻、4°C冷藏或在20°C的环境温度下保存0、24、48、72或96小时),然后在−80°C冷冻。PCoA分析结果表明(图2),受试者差异几乎占一半的总体差异(R^2=0.47 , P< 0.001), 3天内储存条件对口腔菌群组成没有显著性影响(R^2=0.17 , P= 0.2)。
Lauber等人检测了温度和储存时间对土壤、人体皮肤和人类粪便样本中细菌群落相对类群丰度的影响。结果发现细菌群落的总体组成和多数优势类群的相对丰度并未因不同条件而改变(P>0.1)[99]。皮肤和粪便的生物学重复样本是因宿主聚类而不是温度或储存时间。尽管短期内处理样本可以优先考虑便利性,但我们建议样品在采集后立即冷冻,或者使用可替代的储存方法[89]。
低微生物量样本-处理环境污染
与微生物群落丰富的样品(如健康人的粪便)相比,试剂和实验室污染在低微生物量样品中总微生物负荷占比更大。低起始量模板可能被样品处理中的试剂或仪器中的残留的微量DNA所污染,因此部分或全部微生物序列可能来自环境污染。所以,在研究细菌水平较低的身体部位(如人类肺部和皮肤)或正常情况下根本不含任何微生物的部位(如各种健康组织)的微生物菌群时,考虑潜在的污染显得尤为重要[17,19,22]。
Salter等人证明细菌培养物的连续稀释会产生更多的污染序列,随着稀释次数增加,会产生更少的“真实”序列,直到污染序列占总序列的大部分[23]。对于低生物量样本,20个PCR扩增循环太低,但40个循环恢复了污染和真实的低水平序列 [23]。Kennedy报道了起始模板浓度是测序结果差异的主要影响因素[104]。即使在没有PCR扩增步骤的宏基因组样本中,也能检测到少量微生物DNA的样本也会存在类似的污染[23]。
不同试剂盒之间会有差异,甚至在同一试剂盒的不同批次间也会有所不同[20,23]。因此,最好使用相同批次的试剂同时处理所有样品。记录每个样本使用的试剂盒以及批次是至关重要的。如果使用多个试剂盒,应将试剂盒批次作为统计分析中的一个因素。
对于低微生物量样本,我们建议使用专为将污染降至最低而设计的DNA分离试剂盒(如,QIAamp UCP(UltraClean Production)病原体迷你试剂盒(QIAGEN))。因此,如果一个项目同时包含低微生物量样本和高微生物量样本,要为所有样本提供一种试剂盒类型,以避免试剂盒导致的差异。
在分析方面,已经开发了几种方法来过滤可疑的污染分类群。Bittinger等人提出了一种利用真菌总DNA浓度来确定真菌类群来自污染源比例的方法[18],该方法与使用PicoGreen的DNA浓度相似。Lazarevic等人提出了一种结合qPCR计算总DNA浓度的方法,这是一种更准确但更耗费资源的方法[106]。Jervis-Bardy表明,随着总DNA浓度的增加,受污染的分类群相对丰度往往会明显下降,并将此作为另一种去除受污染分类群的方法的依据[21]。我们可以使用SourceTracker对各个污染源进行建模,使用贝叶斯方法来估计来自每个污染源序列数的相对比例[107]。
Lauder从6名人类受试者的胎盘中提取了DNA,并将其与几种类型的空白拭子和只含有试剂的阴性对照一起进行了研究。实时荧光定量PCR定量分析胎盘样本、对照样本和唾液样本(取自同一受试者)的16S rRNA基因拷贝总数,并用两种DNA提取试剂盒进行纯化。无论使用哪种试剂盒,胎盘样本和对照的拷贝数都很低,与阴性对照难以区分,而微生物量较高的唾液样品对两种试剂盒均显示相似的物种组成(图3)。在胎盘和对照样本中发现的几个共同物种是DNA提取试剂盒的已知污染物。由此推断,试剂盒在胎盘样本中提供了主要的微生物特征[22]。未来的研究是否能明确区分胎盘样本和阴性对照,还有待观察。
设置阴性对照
收集阴性对照样本,对污染背景进行评估是十分必要的。我们通常在每个16S rRNA测序反应中设置三种类型的阴性对照样本(图4)。在“空白拭子”样本中,从包装中打开一根无菌棉签,并对其实施完整的测序方案。在“空白提取”样本中,DNA提取和所有后续步骤都是在没有额外加入材料的情况下进行的。在“空白文库”样本中,不实施提取方案;DNA-free water被用在该方案的提取后面步骤中,从文库构建开始,以表征下游步骤中的污染。
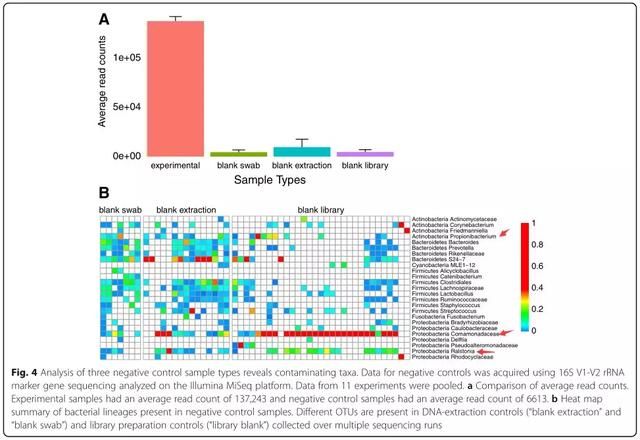
如果微生物量低,可以加入额外的阴性对照样本,以检测样本采集过程中引入的污染DNA。肺部微生物菌群的研究中,在支气管镜检前用灌洗盐水样本冲洗支气管镜可以产生极好的阴性对照[19]。阴性对照样本测序序列平均数量通常比来自粪便等样本的平均数量低5个对数(图4A)。阴性对照样本中发现的细菌类群属于已有文献中报道的污染类群,包括丛毛单胞菌科Comamonadaceae、劳尔氏菌属Ralstonia 和丙酸杆菌属Propionibacterium (图4B)。
设置阳性对照
阳性对照样品可以验证样品制备和测序过程是否顺利进行。当样品在多孔板上纯化时,样品会在板上特定位置进行一致排列,这使得在测序结果中能够跟踪检测到任何样品的混淆。理想情况下,正负对照样品会不对称地放置在提取板上,从而确定板的方向。
大量研究证明,培养的微生物混合物(“模拟群落”)[23,96,111]或已知的游离DNA的混合物(“模拟DNA”样本)[88,112,113]都是有效的阳性对照。测序结果在同一个方法和实验室环境中是可重现的,但是不同方法和实验室可能产生不同误差[23]。
我们合成了八个古菌物种的16S rRNA基因区域,这些区域在实验数据中通常不会被检测到,因为古菌V1-V2区域的扩增引物结合位点的序列与细菌V1-V2引物序列不匹配。我们对古菌样本进行了11次独立测序反应,发现这些序列的相对丰度是基本一致的(图5B)。

图5c显示了来自一次测序反应的代表性结果。在96孔板上,对照古菌类群的丰度并没有因临近阳性对照样品而增加(P=0.6),这表明制备过程中的溢出并不是样品间混杂的主要来源。然而,在多个分散的样本中可以检测到低水平的这些序列(图5C,蓝色正方形),这可能是由于在Illumina测序过程中barcodes的误读或相邻簇中DNA分子的杂交[114]。避免这种情况发生的一种手段是在扩增子两端使用barcodes,并且要求在质量过滤中两端都精确匹配[115]。
gene block是在文库制备和测序过程中保证实验样品适当扩增、跟踪混淆样品和检测样品交叉污染的简单方法。但是,合成的阳性对照是无法用于基准分析和统计方法的。由于序列的自然变异和低丰度类群导致为真实群落开发的分析方法通常在模拟群落上表现不佳,反之亦然。
许多研究者使用能同时靶向细菌和古菌16S区域的引物,例如地球微生物组项目使用的515fB/806rB引物[116,117]。因此,在gene block中使用古生菌序列没有优势,因为在实验样本中可能检测到古生菌。尽管如此,研究人员可以使用人工改变的DNA序列来构建gene block,这些DNA序列有足够可变性,可以很好地与基因组序列区分开来,但又足够保守,可以与分析流程兼容。
鸟枪法宏基因组测序中的污染
鸟枪法宏基因组测序中也可以检测到由试剂引入的微生物DNA。而扩增子测序,在微生物生物量较低的样品中污染尤为明显。这在微生物量普遍较低的样品(如皮肤拭子)和以非微生物DNA为主的样本(如组织活检)中都可以看到。

我们对使用标准试剂盒(DNasy PowerSoil,Qiagen,Valencia,CA,USA)和低污染试剂盒(QIAamp UCP Pathogen,Qiagen,Valencia,CA,USA)提取的组织DNA进行了鸟枪法宏基因组测序(未发表的数据)。对阴性对照样本进行测序时,我们观察到两种试剂盒检测的菌群不同(图6A)。这两种试剂盒中发现的菌群在低微生物量组织样本中也存在,很可能是来自试剂污染。在实验和对照样本中发现的菌群包括丙酸杆菌属、棒状杆菌和Bradyrhyzobium(常见的土壤细菌),也被其他研究组鉴定为污染物[23,118]。
这表明,尽管一些试剂污染是不可避免的,但使用低污染试剂盒减少了污染物引起的的总测序量。此外,它还强调了测序和分析对照样本的重要性,如果没有它们,就不可能从真正微生物菌群里鉴定试剂污染。
检测污染的一个极端例子来自病毒分析,使用了多重置换扩增技术来扩增样本。对空白的病毒样本(未发表的数据)进行鸟枪法宏基因组测序,并与噬菌体phi29比对,发现比对结果仅与phi29聚合酶基因一致(图6B)。
三、分析中注意的问题
处理阴性对照
与其他样品一样,检测阴性对照样品的组成是必要的。即使文库产率低或检测不到,也要对阴性对照进行测序,使用堆叠条形图或热图来展示其组成。不要认为只要在阴性对照中去掉污染物种,污染问题就已经解决。我们没有理由认为污染的物种是完全被抽检的,而且环境物种可能确实存在于样本中,并且具有重要的功能。
多重比较控制显著性和假阳性率
高通量测序实验通常会产生来自数百个分类群的测序序列。研究者要想知道哪些分类群可能与表型差异有关,就必须进行多重比较,每次都要检验分类群的丰度没有差异的零假设。如果检验的可接受假阳性率被设置在某个水平(如5%),则这些重复比较将增加获得高于该水平的假阳性的机会。想要将假阳性率重新调整回要求水平,必须使用多重检验进行校正。
一种保守的方法是使用Bonferroni校正[120]来确保在指定的概率内没有任何假设被错误地拒绝。但这种方法已经被证明是不可接受的,会导致太多的假阴性。另一种更流行的方法是控制预先指定的错误发现速率(即对零假设的错误拒绝)。Benjamini和Hochberg提出了一种在一系列独立检验中控制错误发现率的方法[121],这是QIIME[122]和Mothur[123]等微生物组分析软件中使用的方法。我们强烈建议在进行多重比较时要进行多重检验校正。
发现队列和验证队列双重检验提高结果可信度
微生物组研究中,首先在发现队列中进行实验,使用特定的检验方法筛选分类群或基因型,然后对验证队列进行分析来检验仅在发现队列中显著的结果。这样,验证队列中的检验总数就大大减少了。
一些微生物组研究已经使用独立的发现和验证队列来筛选与疾病状态相关的生物标志物。Sabino等人在一个发现队列中鉴定了三个与原发性硬化胆管炎相关的菌属,并在独立验证队列中将75%的受试者进行了正确分类[125]。Forslund等人使用独立的队列来重复在二甲双胍治疗的2型糖尿病患者中发现的分类群改变的结果[126]。在一组受试者中开发了与炎症性肠病(IBD)相关的粪便细菌分类群丰度的综合指数[73],然后在一项独立的随访研究中发现,该指数可以将IBD与健康对照组区分开来[127]。Kelsen 等人应用发现—验证队列设计来确定克罗恩病儿童和健康对照组之间龈下微生物菌群的差异[128]。此外,他们能够将与抗生素使用相关的分类群与仅与疾病相关的分类群区分开来。
四、结论
我们对微生物组研究的实验设计和分析提出以下几点建议:
需要考虑多个混杂因素,包括抗生素的使用、年龄、性别、饮食、地理和宠物饲养。
在多个笼子中设置要研究的条件是十分重要的,这样可以隔离和解释笼子变量的效应。
虽然我们建议在采集后立即将样品(特别是粪便样品)保存在−80°C以获得最准确的结果,但现场研究的替代存储方法也会导致相对较小的偏差。对于新的样品类型,特定的存储条件最好要检测存储过程中的菌群变化。
在横断面研究中,了解取样的时间点是否具有代表性是至关重要的。例如,健康成人的肠道菌群在短期内不会发生根本性的变化,但阴道菌群有时会发生变化。因此,评估可能的纵向变化与所提问题之间的关系是很重要的。
要认真地设置和分析阴性对照:DNA提取试剂盒通常带有污染物,不同试剂盒甚至同一试剂盒不同批次间的污染也可能会有所不同。
每批样品设置阳性对照。模拟群落是非常有用的,我们给出了可用于阳性对照的DNA合成序列列表。在纯化板上不对称地设置对照样品,通过DNA纯化和文库制备程序来验证对样品的跟踪是否准确。
低微生物量样品面临众多挑战。进行低微生物量样本研究时,我们可以采用qPCR来量化样本中的微生物负荷,以了解实验困难的程度。在这种实验中,先从所有序列数据只反映存在污染的零假设开始,然后询问在数据的统计分析中是否可以拒绝这个假设。
对“数据挖掘”要实事求是,对多重比较要严格控制。
条件允许的话,最好通过发现和验证队列进行双重检验来提高实验的稳健性。
毫无疑问,人类微生物菌群对健康和疾病至关重要。通过应对上述挑战,我们可以获得高质量的数据,从而促进微生物组研究领域的快速发展。
参考文献:
Kim Dorothy, Hofstaedter Casey E, Zhao Chunyu et al. Optimizing methods and dodging pitfalls in microbiome research [J]. Microbiome, 2017, 5: 52. DOI:10.1186/s40168-017-0267-5.
如果您想了解更多关于微生物组研究的知识和前沿进展,可以扫描下方二维码申请加入欧易-微生物测序交流 QQ 群( 群号:598662347), 群内会推荐微生物组研究领域最新文献,举办微生物组研究系列讲座。

我们在这里等你哟, 一起看文献, 学知识, 做科研!
END
本文系欧易生物原创
转载请注明本文转自欧易生物