WGCNA算法研究笔记
文章的作者将该算法用R实现,正好数模过后我也在进行matlab向R的转换,这套算法就成为了我自学R的良好素材(TIOBE今年三月公布的编程语言排行榜中,R列居第24位,超过了SAS和Matlab,看来自己的选择似乎不错)。
关于该实例的数据和分析说明可以在以下网页中找到 http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/Rpackages/WGCNA/ 由于我不想写成一篇翻译稿,因此只会点到代码中的一些关键环节,并解释每一步分析的作用。
WGCNA所给的样本数据是F2代135只小鼠liver细胞的芯片杂交结果,需要以此进行module identification,并将建立好的module与性状关联。
(1)数据的预处理:
# Display the current working directory
getwd();
# If necessary, change the path below to the directory where the data files are stored.
# 指定到数据所在目录
workingDir = ".";
setwd(workingDir);
# Load the package
# 这里需要保证计算机的R中安装了WGCNA这一分析包
library(WGCNA);
# The following setting is important, do not omit.
options(stringsAsFactors = FALSE);
#Read in the female liver data set
femData = read.csv("LiverFemale3600.csv");
# Take a quick look at what is in the data set:
dim(femData);
names(femData);
# 从样本数据中抽取表达量数据
# 有一点值得注意的是,样本数据中芯片表达量存在负值,为此我纠结可很长时间
# 事实上,芯片表达数据在给出时是相对一个参比而言的,即将目的基因与参比基因的表达量相除
# 然后取其对数值,得到每一个基因的表达量,因此如果目的基因的表达量低于参比基因
# 那么其数据值将出现负数
datExpr0 = as.data.frame(t(femData[, -c(1:8)]));
names(datExpr0) = femData$substanceBXH;
rownames(datExpr0) = names(femData)[-c(1:8)];
# 检测异常值,个人觉得没什么太大用处,所有的基因都通过了检验
gsg = goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK
if (!gsg$allOK)
{
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# Remove the offending genes and samples from the data:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
调用聚类函数,依据不同小鼠芯片的表达情况,对小鼠进行聚类,并作图
sampleTree = flashClust(dist(datExpr0), method = "average");
# Plot the sample tree: Open a graphic output window of size 12 by 9 inches
# The user should change the dimensions if the window is too large or too small.
sizeGrWindow(12,9)
#pdf(file = "Plots/sampleClustering.pdf", width = 12, height = 9);
par(cex = 0.6);
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
# 插入聚类分割线
# Plot a line to show the cut
abline(h = 15, col = "red");
# Determine cluster under the line
clust = cutreeStatic(sampleTree, cutHeight = 15, minSize = 10)
table(clust)
# clust 1 contains the samples we want to keep.
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
# 导入性状信息
traitData = read.csv("ClinicalTraits.csv");
dim(traitData)
names(traitData)
# remove columns that hold information we do not need.
# 提取有用的性状信息
allTraits = traitData[, -c(31, 16)];
allTraits = allTraits[, c(2, 11:36) ];
dim(allTraits)
names(allTraits)
# Form a data frame analogous to expression data that will hold the clinical traits.
femaleSamples = rownames(datExpr);
traitRows = match(femaleSamples, allTraits$Mice);
datTraits = allTraits[traitRows, -1];
rownames(datTraits) = allTraits[traitRows, 1];
collectGarbage();
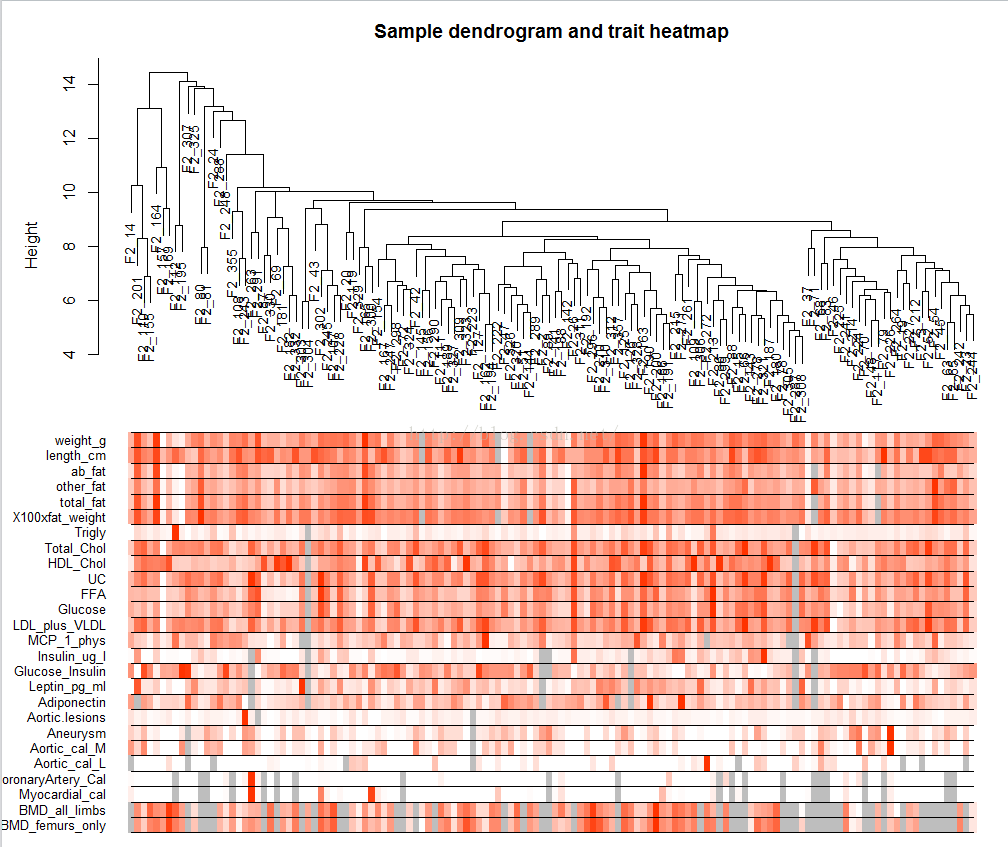
# 将小鼠的性状量化值在聚类图中表示出来
# Re-cluster samples
sampleTree2 = flashClust(dist(datExpr), method = "average")
# Convert traits to a color representation: white means low, red means high, grey means missing entry
traitColors = numbers2colors(datTraits, signed = FALSE);
# Plot the sample dendrogram and the colors underneath.
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
# 保存处理好的表达信息和性状信息
save(datExpr, datTraits, file = "FemaleLiver-01-dataInput.RData")
下图的上半部分是将135只小鼠按照其基因表达的相似性进行聚类的结果,下半部分为性状的可视化,采用heatmap的形式,将135只老鼠的性状量化值表示出来,颜色越红,其数值越大。
(2)网络的建立:
为了简化这里采用automatic的方法。
# 首先导入(1)步骤预处理好的数据
# Display the current working directory
getwd();
# If necessary, change the path below to the directory where the data files are stored.
# "." means current directory. On Windows use a forward slash / instead of the usual \.
workingDir = ".";
setwd(workingDir);
# Load the WGCNA package
library(WGCNA)
# The following setting is important, do not omit.
options(stringsAsFactors = FALSE);
# Load the data saved in the first part
lnames = load(file = "FemaleLiver-01-dataInput.RData");
#The variable lnames contains the names of loaded variables.
lnames
# 选择一系列的power参数进行网络构建
# 根据topology overlap criteria选择最佳的参数进行进一步分析
# Choose a set of soft-thresholding powers
powers = c(c(1:10), seq(from = 12, to=20, by=2))
# Call the network topology analysis function
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
# Plot the results:
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
# 选择好参数后,建立网络
net = blockwiseModules(datExpr, power = 6, minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
从图中可以看出回归系数 值与基因的连接数间的trade-off,随着power值得增加, 值不断增加,表明网络越接近于scale-free network,但是基因的连接数减少使得module identification逐渐失去意义。因此该问题有待进一步研究,建立一优化模型不失为一个好的选择。
# 显示每一个module的基因数
table(net$colors)
# 绘制出基因的module分类图
# open a graphics window
sizeGrWindow(12, 9)
# Convert labels to colors for plotting
mergedColors = labels2colors(net$colors)
# Plot the dendrogram and the module colors underneath
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
如图,对芯片中所有基因按照TOM矩阵值进行聚类的结果,module color中每一种颜色对应聚类树上的基因属于一个module。
# 保存识别好的module信息
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(MEs, moduleLabels, moduleColors, geneTree,
file = "FemaleLiver-02-networkConstruction-auto.RData")