在实践之前,我先查找了一下有关组蛋白修饰的知识点:
(参考文章:https://www.jianshu.com/p/8aca72809c5c)
组蛋白修饰能预测染色质的类型(异染色质或常染色质)、区分基因组功能元件(启动子、增强子、基因主体)以及检测决定这些元件处于活性状态或是抑制状态。例如H3K4me2和H3K4me3修饰大多数富集在转录起始位点附近的启动子上激活基因表达,而H3K27me2和H3K27me3与基因抑制相关。因此可通过CHIP-seq分析组蛋白修饰的分布寻找基因的启动子区和增强子区域及其是激活或抑制基因表达。
H3K4me1可作为增强子的标志,H3K4me3作为启动子标志。研究表明,H3K4me1和H3K4me3与基因激活相关,H3K4me3主要富集在转录起始位点附近的启动子区域,而大多数H3K4me1修饰富集在增强子区域;H3K27ac与基因激活相关,主要富集在增强子和启动子区域,当增强子区只有H3K4me1修饰富集时,该增强子处于平衡状态,而当增强子区域同时富集H3K4me1和H3K27ac修饰时,该增强子就处于激活状态促进基因表达;H3K27的甲基化是可逆的过程,H3K27me1显示出对转录具有正向影响,启动子区域的H3K27me3甲基化修饰时抑制基因的转录,而H3K27me2广泛分布并且在沉默非细胞类型特异性增强子中起作用。
下表为常见的组蛋白修饰的主要分布及功能:

异染色质是染色质的浓缩,转录无活性状态,H3K9甲基化是异染色质的标志。H3K27me1和H3K9me3存在于着丝粒异染色质区域,而H3K27me3和H3K9me2共同存在于抑制的常染色质区域中。H3K9ac也与H3K14ac和H3K4me3高度共存共同作为活性基因启动子的标志。
本次实践文章:
Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma
用不同浓度的OSCC抗癌物THZ1处理两种OSCC细胞系:KYSE510和TE7。
寻找药物处理后的超级增强子。
1.下载SRA原始数据:
#!/bin/bash
for ((i=251;i<=254;i++))
do wget https://sra-download.ncbi.nlm.nih.gov/traces/sra28/SRR/003028/SRR3101$i
done
2.提取fastq文件:
#!/bin/bash
for i in SRR310125*
do
echo $i
fastq-dump --gzip --split-files $i
done
每运行一个文件,都会在家目录下的ncbi文件里生成一个临时文件,非常占内存。由于我的存储空间没有了,所以我就直接下载了fastq文件。下载fastq文件的网址是:https://www.ebi.ac.uk/ena/data/view/SRX1530372。再复制下fastq.gz文件的地址。
网上有的文章提到:尽量不要用wget或curl去下载sra文件,某些情况下会导致下载的文件不完整。所以这次我也尝试了用ascp下载的方法。
这里说一下用ascp下载的方法。
安装ASCP:
#下载
$ wget http://download.asperasoft.com/download/sw/connect/3.7.4/aspera-connect-3.7.4.147727-linux-64.tar.gz
$ tar zxvf aspera-connect-3.7.4.147727-linux-64.tar.gz
#安装
$ bash aspera-connect-3.7.4.147727-linux-64.sh
#要去home目录里查看是否有.aspera文件夹
$ cd /home
$ ls -a
#如果看到.aspera文件夹,代表安装成功
#永久添加环境变量
$ echo 'export PATH=~/.aspera/connect/bin:$PATH' >> ~/.bashrc
$ source ~/.bashrc
#查看是否可以调用
$ ascp --help
ascp的用法:ascp [参数] 目标文件 目标地址,在线文档
ascp命令的常用参数:
-v verbose mode 唠叨模式,能让你实时知道程序在干啥,方便查错。
-T 取消加密,否则有时候数据下载不了。
-i 提供私钥文件的地址。
-l 设置最大传输速度,一般200m到500m,如果不设置,反而速度会比较低,可能有个较低的默认值。
-k 断点续传,一般设置为值1
-Q 一般加上它
-P 提供SSH port,一般是33001。
在EBI网站copy下载地址一般是ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR310/004/SRR3101254/SRR3101254.fastq.gz这样的格式。需要把ftp://ftp.sra.ebi.ac.uk换成[email protected]:。NOTE: 这篇文章的ChIP-seq是单端测序。
我的代码:
$ ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/vol1/fastq/SRR310/001/SRR3101251/SRR3101251.fastq.gz .
$ ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/vol1/fastq/SRR310/002/SRR3101252/SRR3101252.fastq.gz .
$ ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/vol1/fastq/SRR310/003/SRR3101253/SRR3101253.fastq.gz .
$ ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/vol1/fastq/SRR310/004/SRR3101254/SRR3101254.fastq.gz .
另外要注意的是:[email protected]后面是:号,不是路径
最后gz结尾有一个空格,然后是"."。如果没有空格和点,则会报错。无法下载。
3.使用fastqc质量检查
4.bowtie2比对
下面这些代码是按照上一次实践的方法写的,直接生成bam文件。因为这篇文献的测序结果质量很好,所以没有去掉3'端的几个碱基
#!/bin/bash
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101251.fastq.gz | samtools sort -O bam -o /media/yanfang/FYWD/CHIPSEQ/sambam/TE7_H3K27Ac.bam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101252.fastq.gz | samtools sort -O bam -o /media/yanfang/FYWD/CHIPSEQ/sambam/TE7_input.bam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101253.fastq.gz | samtools sort -O bam -o /media/yanfang/FYWD/CHIPSEQ/sambam/KYSE510_H3K27Ac.bam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101254.fastq.gz | samtools sort -O bam -o /media/yanfang/FYWD/CHIPSEQ/sambam/KYSE510_input.bam
下面这些代码是bowtie2比对后生成sam文件的方法。有些文章里在进行比对的时候是用的bowtie进行比对的,并且设置了只保留比对一次的reads。我这里用的是bowtie2,所以没有设置这个参数。
#!/bin/bash
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101251.fastq.gz -S TE7_H3K27Ac.sam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101252.fastq.gz -S TE7_input.sam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101253.fastq.gz -S KYSE510_H3K27Ac.sam
bowtie2 -p 6 -x /media/yanfang/FYWD/CHIPSEQ/fqfile/bowtie2hg19/hg19 -U /media/yanfang/FYWD/CHIPSEQ/fqfile/SRR3101254.fastq.gz -S KYSE510_input.sam
#比对率如下:
64273075 reads; of these:
64273075 (100.00%) were unpaired; of these:
1285733 (2.00%) aligned 0 times
46680241 (72.63%) aligned exactly 1 time
16307101 (25.37%) aligned >1 times
98.00% overall alignment rate
[bam_sort_core] merging from 16 files and 1 in-memory blocks...
64402758 reads; of these:
64402758 (100.00%) were unpaired; of these:
1271927 (1.97%) aligned 0 times
45320875 (70.37%) aligned exactly 1 time
17809956 (27.65%) aligned >1 times
98.03% overall alignment rate
[bam_sort_core] merging from 16 files and 1 in-memory blocks...
64555367 reads; of these:
64555367 (100.00%) were unpaired; of these:
901311 (1.40%) aligned 0 times
48336949 (74.88%) aligned exactly 1 time
15317107 (23.73%) aligned >1 times
98.60% overall alignment rate
[bam_sort_core] merging from 16 files and 1 in-memory blocks...
68947224 reads; of these:
68947224 (100.00%) were unpaired; of these:
4955211 (7.19%) aligned 0 times
45955461 (66.65%) aligned exactly 1 time
18036552 (26.16%) aligned >1 times
92.81% overall alignment rate
[bam_sort_core] merging from 17 files and 1 in-memory blocks...
5.使用MACS获得Chip-seq富集区
以下是官方网站对于常规峰和“宽”峰的一些解释(https://www.encodeproject.org/chip-seq/histone/) 。有的文章说常规峰和宽峰分析过程会有些不一样,所以我特异查了一下H3K27Ac是什么峰,根据这个表来看属于常规峰。
- For narrow-peak histone experiments, each replicate should have 20 million usable fragments.
- For broad-peak histone experiments, each replicate should have 45 million usable fragments.
- H3K9me3 is an exception as it is enriched in repetitive regions of the genome. Compared to other broad marks, there are few H3K9me3 peaks in non-repetitive regions of the genome in tissues and primary cells. This results in many ChIP-seq reads that map to a non-unique position in the genome. Tissues and primary cells should have 45 million total mapped reads per replicate.
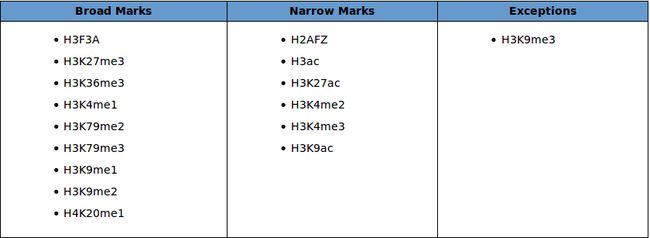
- For narrow-peak histone experiments, each replicate should have 10 million usable fragments(https://www.encodeproject.org/data-standards/terms/#read-depth).
- For broad-peak histone experiments, each replicate should have 20 million usable fragments.
$ macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/TE7_input.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/TE7_H3K27Ac.bam -m 10 30 -p 1e-5 -f BAM -g hs -n TE7_H3K27Ac 2>TE7_H3K27Ac.masc2.log
$ macs2 callpeak -c /media/yanfang/FYWD/CHIPSEQ/sambam/KYSE510_input.bam -t /media/yanfang/FYWD/CHIPSEQ/sambam/KYSE510_H3K27Ac.bam -m 10 30 -p 1e-5 -f BAM -g hs -n KYSE510_H3K27Ac 2>KYSE510_H3K27Ac.masc2.log
-c:对照组。
-t:处理组。
--format:输入的文件类型。
--输出的文件前缀名称。
--keep-dup对于重复序列的处理方式,1效果最好。指明MACS在染色体同一位置的reads(重复序列处理方式)。
--wig:输出的文件类型。
--single-profile表示输出单文件。
--space是文献中的要求。(Wiggle files were generated using read pileups for every 50 base pair bins)
--m:This parameter is used to select the regions within MFOLD range of high-confidence enrichment ratio against background to build model. The regions must be lower than upper limit, and higher than the lower limit of fold enrichment. DEFAULT:10,30 means using all regions not too low (>10) and not too high (<30) to build paired-peaks model. If MACS can not find more than 100 regions to build model, it will use the --shiftsize parameter to continue the peak detection.This parameter is used to select the regions within MFOLD range of high-confidence enrichment ratio against background to build model. The regions must be lower than upper limit, and higher than the lower limit of fold enrichment. 构建双峰模型时使用,默认[10,30]。表示选择那些倍数变化在10-30之间的富集区域,如果找不到100个区域构建模型,并且你还设置了--fix-biomodal,它就会用--shiftsize参数继续分析。
call peak 后得到以下几个文件类型:

打开peak.xls文件后,表头有几行显示:

6.构建index文件
为了在IGV里查看peak,需要把bam文件先构建index,即生成bam.bai文件:
$ samtools index TE7_H3K27Ac.bam TE7_H3K27Ac.bam.bai
$ samtools index TE7_input.bam TE7_input.bam.bai
$ samtools index KYSE510_H3K27Ac.bam KYSE510_H3K27Ac.bam.bai
$ samtools index KYSE510_input.bam KYSE510_input.bam.bai
顺便插一句:[生信人必会之samtools](https://vip.biotrainee.com/d/117-samtools)
这篇文章大概是我看到过写的最好最全面的samtool使用方法。
7.用deeptools里的bamcoverage工具将bam文件标准化
$ bamCoverage -b TE7_H3K27Ac.bam -o TE7_H3K27Ac.bw -p 10 --normalizeUsing RPKM
$ bamCoverage -b TE7_input.bam -o TE7_input.bw -p 10 --normalizeUsing RPKM
$ bamCoverage -b KYSE510_H3K27Ac.bam -o KYSE510_H3K27Ac.bw -p 10 --normalizeUsing RPKM
$ bamCoverage -b KYSE510_input.bam -o KYSE510_input.bw -p 10 --normalizeUsing RPKM
将生成的bw文件(标准化后的)导入IGV:




其他文献里涉及到的IGV的图,都几乎和我做出来的一模一样,说明分析过程基本没啥问题。
7.ROSE筛选super-enhancer
这部分是本次实践的重点,ROSE官网:http://younglab.wi.mit.edu/super_enhancer_code.html
有关这个ROSE软件讲解的比较好的中文版的文章:https://blog.csdn.net/oxygenjing/article/details/77862115
ROSE的最主要用法有ROSE_main.py和ROSE_geneMapper.py。其中ROSE_main.py 用于寻找增强子而ROSE_geneMapper.py 用于为增强子相关的基因进行注释。
首先看一下ROSE_mian.py 用法:
(1)安装ROSE,我们直接从其托管在Bitbucket仓库中克隆Python脚本
git clone https://bitbucket.org/young_computation/rose.git
也可以像这篇文章一样https://blog.csdn.net/oxygenjing/article/details/77862115,安装ROSE
$ wget https://bitbucket.org/young_computation/rose/get/1a9bb86b5464.zip
$ unzip 1a9bb86b5464.zip
(2)安装后,首先准备文件:peaks的gff文件。这个peak文件是MACS2 call peak之后生成的peaks文件。
官方网站对gff文件的格式要求:
.gff must have the following columns:
1: chromosome (chr#)
2: unique ID for each constituent enhancer region
4: start of constituent
5: end of constituent
7: strand (+,-,.)
9: unique ID for each constituent enhancer region
所以要先从peak文件里提取这些要用的列。这里要注意的是:1,2,4,5,7,9是gff文件的列数,第3,6,8列不是不要了,而是要都写成“ ”空格形式。
(刚开始我以为其他三列都不要了,所以在gff文件里只有6列,结果一直报错。后来按照某篇教程里写的其他三列都换成".",结果也一直报错......整整折腾了我3天的时间,死活跑不通。)
$ awk '{print $1"\t"$10"\t"" ""\t"$2"\t"$3"\t"" ""\t"".""\t"" ""\t"$10}' TE7_H3K27Ac_peaks.bed > TE7_peaks.gff
这里的\t是以制表符分割的意思。每一列用\t分割,并且用""括起来。
(3)接下来创建一个虚拟的python2.7的环境,因为我装的是python3,所以需要创建一个虚拟的环境来运行ROSE。
source activate py27
(4)还需要将samtools的PATH设置到这个文件夹里,因为ROSE运行时需要调用samtools。如果你的samtool没有设置全局变量,是要多一步添加PATH的。但是因为我的samtool已经在全局变量里,可以直接在这个文件夹里打"samtools"就可以调用,所以我就没有再设置环境变量了。
(5)之后我们需要bam文件(这个bam文件是需要先sort的。处理组,对照组),gff文件(处理组),bam.bai文件(也就是bam的index文件,处理组和对照组)。把这些文件全都放进ROSE安装的文件夹里,切记:运行ROSE的时候也要在其安装的文件夹里运行,我也不知道这软件开发的人是咋想的。。。
然后一切准备好之后,就是见证奇迹的时刻了。(虽然我这个奇迹到了第4天才看到。。。之前3.9天都在解决报错问题了)
python ROSE_main.py -g HG19 -i ./TE7/TE7_peaks.gff -r ./TE7/TE7_H3K27Ac_sorted.bam -c ./TE7/TE7_input_sorted.bam -o roseresult -s 12500 -t 2500
如果运行时有报错,例如有类似如下显示:
Traceback (most recent call last):
File "ROSE_bamToGFF.py", line 249, in
main()
File "ROSE_bamToGFF.py", line 240, in main
newGFF = mapBamToGFF(bamFile,gffFile,options.sense,int(options.extension),options.floor,options.rpm,options.matrix)
File "ROSE_bamToGFF.py", line 76, in mapBamToGFF
gffLocus = ROSE_utils.Locus(line[0],int(line[3]),int(line[4]),line[6],line[1])
请确保你的bam文件格式,gff文件格式,samtool的环境变量都没有问题的情况下,并且所有需要的文件在ROSE安装的文件夹里,仍然出现这些报错,你可以尝试这样修改源代码:(参考官方给出的解决方法:https://bitbucket.org/young_computation/rose/issues/44/typeerror-issue)
FIX:
Make the following changes to ROSE_utils.py:
Line 602 (original): command = 'samtools view %s | head -n 1' % (bamFile)
Line 602 (new): command = './samtools view %s | head -n 1' % (bamFile)
Line 632 (original): command = 'samtools flagstat %s' % (self._bam)
Line 632 (new): command = './samtools flagstat %s' % (self._bam)
Line 657 (original): command = 'samtools view %s %s' % (self._bam,locusLine)
Line 657 (new): command = './samtools view %s %s' % (self._bam,locusLine)
最后运行后会生成很多文件:
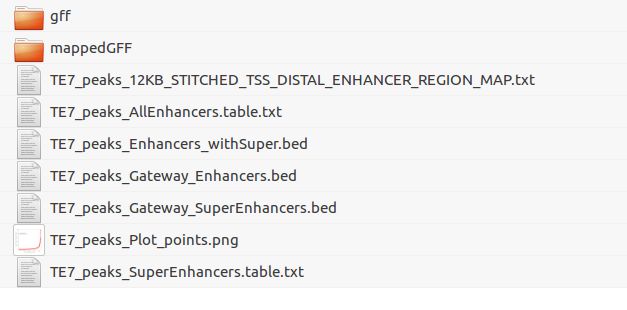
输出文件如下(参考文章:https://blog.csdn.net/oxygenjing/article/details/77862115):
1.gtf(这个文件在gff文件夹里): 该文件为输入gtf文件的副本。
2.stitched.gtf(这个文件也在gff文件夹里):该文件为通过STITCHING_DISTANCE将INPUT_CONSTITUENT_GFF拼接在一起创建的gff文件;文件列数如下:
chrom, name, [blank], start, end, [blank], [blank], strand, [blank], [blank], name
其中 name 字段的命名方式为:拼接起来的区域数+最左端区域ID。
3._MAPPED.gff(在mappedGFF文件夹里): 每个bam文件通过bamToGFF的输出文件,包含以下列:
(成分ID,测试区域,平均读取密度(单位为每百万位元每百万映射的单位读数密度))
4.* _STITCHED * _MAPPED.gff(在mappedGFF文件夹里,处理组和对照组分别有一个文件): 每个bam文件通过bamToGFF的输出文件,该文件中对增强子区域进行了拼接,包含以下列:
(拼接增强子ID,测试区域,平均读取密度(单位为百万映射每单位拼接增强子数))
5.STITCHED_ENHANCER_REGION_MAP.txt: 利用bamToGFF计算后得到的拼接enhancer密度文件,包含以下列:
(拼接增强子ID,染色体,拼接增强子起始位置,拼接增强子末端位置,拼接数,BAM信号等级,BAM信号)
- AllEnhancers.table.txt:enhancer列表,包含每个增强子的排名和是否为超级增强子,内容包括:增强子ID,染色体,拼接增强子起始位点,拼接增强子末端,拼接数,拼接成分大小,BAM的信号,BAM的等级,是否为超增强子:是(1)否(0)
7._SuperEnhancers.table.txt: 超级enhancer的排名,为_AllEnhancers.table.txt 文件的子集。包含以下列:
(拼接增强剂ID,染色体,拼接增强子起始位点,拼接增强子末端,拼接数,缝合在一起的成分的大小,RANKING_BAM的信号,RANKING_BAM的等级,超增强子的二进制(1)与典型(0))
8._Enhancers_withSuper.bed 可以加载到UCSC浏览器中可视化的enhancer bed文件
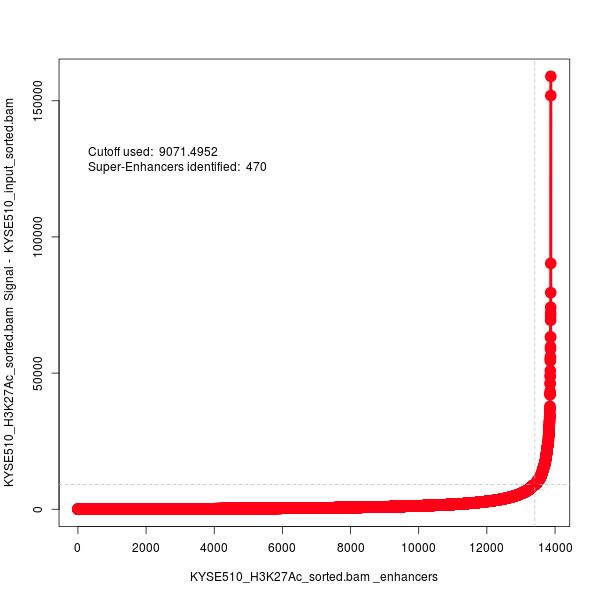
其中那个png图片就是我们需要的:所有enhancer的散点图

之后是KYSE510细胞系的:
python ROSE_main.py -g HG19 -i ./KYSE510_peaks.gff -r ./KYSE510_H3K27Ac_sorted.bam -c ./KYSE510_input_sorted.bam -o KYSE510_roseresult -s 12500 -t 2500
8.ROSE注释enhancer
python ROSE_geneMapper.py -i ./TE7_roseresult/TE7_peaks_AllEnhancers.table.txt -g HG19 -o ./TE7_roseresult 2> ./TE7_roseresult/TE7_enhancer_anno.log
python ROSE_geneMapper.py -i ./KYSE510_roseresult/KYSE510_peaks_AllEnhancers.table.txt -g HG19 -o ./KYSE510_roseresult 2> ./KYSE510_roseresult/KYSE510_enhancer_anno.log
得到以下两个表格:
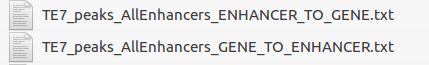
1.ENHANCER_TO_GENE.txt: enhancer重叠基因、附近基因以及最近的基因列表
2.GENE_TO_ENHANCER.txt: 以每个基因为列名的和其相关的增强子位置信息列表(https://blog.csdn.net/oxygenjing/article/details/77862115)
9.利用R做GO富集分析
这部分我也是新手,不知道过程是否正确,反正是跑通了,对不对的还需再多看看。。。
首先在上一步里,我得到了_peaks_AllEnhancers_GENE_TO_ENHANCER.txt这个表,我用的这个表进行的GO分析。
>library(clusterProfiler)
>library(DOSE)
>library(org.Hs.eg.db)
#TE7的enhancer GO富集分析
#首先读取文件,第一行作为列名
>enhancer.relate.gene<-read.table(file="TE7_peaks_AllEnhancers_GENE_TO_ENHANCER.txt",header=T)
#因为在这个txt里的最后一列注明是否为super enhancer,如果是则为1;如果不是则空白。所以取为1的所有行,即挑选出所有的super enhancer
>gene<-enhancer.relate.gene[which(enhancer.relate.gene$IS_SUPER=='1'),]
#查看super enhancer的数据集
> head(gene)
GENE_NAME REFSEQ_ID PROXIMAL_ENHANCERS ENHANCER_RANKS IS_SUPER
1 LAMA5 NM_005560 chr20:60924936-60956028 1 1
2 RPS21 NM_001024 chr20:60924936-60956028 1 1
3 C20orf151 NM_080833 chr20:60924936-60956028 1 1
4 ADRM1 NM_007002 chr20:60924936-60956028 1 1
5 CABLES2 NM_031215 chr20:60924936-60956028 1 1
6 MIR205 NR_029622 chr1:209528209-209591314,chr1:209607925-209609032 2,1441 1
#将这个super enhancer的table第一列提出来,因为第一列是基因名。
gene_name<- gene[,1]
#查看基因名
head(gene_name)
[1] "LAMA5" "RPS21" "C20orf151" "ADRM1" "CABLES2" "MIR205"
#调用R包
library("stringr")
library("clusterProfiler")
#GO的BP分析
ego_bp <- enrichGO(gene_name,
OrgDb = org.Hs.eg.db,
keyType = 'SYMBOL',
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.001,
)
#泡泡图
dotplot(ego_bp, font.size=10)

#GO图
plotGOgraph(ego_bp)

#bar图
barplot(ego_bp,showCategory = 10,title="The GO_BP enrichment analysis of all super enhancers-related genes in TE7")+
scale_size(range=c(2, 12))+
scale_x_discrete(labels=function(ego_bp) str_wrap(ego_bp,width = 25))

KYSE510的分析过程和结果我就不放进来了,只需要把输入的文件换掉就行,其他的一模一样。