246.三元图的应用与绘图实战
三元图在微生物组研究中的应用
本节作者:吴一磊,中科院微生物所
版本1.0.5,更新日期:2020年6月28日
本项目永久地址:https://github.com/YongxinLiu/MicrobiomeStatPlot ,本节目录 246TernaryPlot,包含R markdown(*.Rmd)、Word(*.docx)文档、测试数据和结果图表,欢迎广大同行帮忙审核校对、并提修改意见。提交反馈的三种方式:1. 公众号文章下方留言;2. 下载Word文档使用审阅模式修改和批注后,发送至微信(meta-genomics)或邮件([email protected]);3. 在Github中的Rmd文档直接修改并提交Issue。审稿人请在创作者登记表 https://www.kdocs.cn/l/c7CGfv9Xc 中记录个人信息、时间和贡献,以免专著发表时遗漏。
背景知识
三元图(Ternary plot)是描述三个变量之和为常数的质心图,其核心原理是:
等边三角形内任意一点到三角形三边的距离之和等于其中一边上的高(常数);
过等边三角形内任意一点分别向三条边作平行线,按顺时针方向或逆时针方向读取平行线在各边所截取之三条线段,三条线段之和等于该等边三角形任一边之长(常数)。
在微生物组领域,我们主要利用的是基于相位网格的三元图,其特点是每一种成分在相应等边三角形的顶点的比例为100%,在其对面的线为0%,将零点线和顶点按比例划分,用以估计各成分的含量。如图1:y对面的底线代表落在底线上的所有点在y中占比为0,随着平行线逐渐靠近顶点,落在相应平行线上的点在y中的占比越来越高。
if (!requireNamespace("ggtern", quietly=T))
install.packages("ggtern")
library(ggtern)
data <- data.frame(value=c(10, 30, 50, 70, 100),
x=c(90, 70, 50, 30 ,10),
y= c(10, 30, 50, 70, 90),
z=rep(0, 5))
lables <- data.frame(value=c(10, 30, 50, 70, 90),
x=rep(0.5, 5),
y=c(0.2, 0.35, 0.5, 0.65, 0.8))
p=ggtern(data,aes(x,y,z))
p=p + geom_crosshair_tern() + geom_mask() +
geom_text_viewport(x=lables$x, y=lables$y, label=lables$value, color='red') +
theme_bw()
p
# 保存位图和矢量图,分别用于预览和排版
ggsave(paste0("b1.TenaryPlot.png"), p, width=89, height=89, units="mm")
ggsave(paste0("b1.TenaryPlot.pdf"), p, width=89, height=89, units="mm")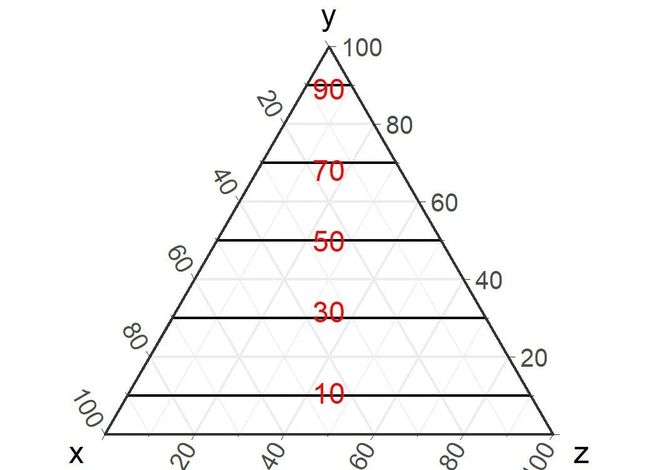
图1. 在y组中的占比逐渐增长
在解读三元图时,我们可以通过点的位置快速获得其在三个分组中的相对比例信息:目标点越靠近一角的顶点,说明他在相应的分组中比例越高;反之其相对比例就越低。根据图1,经过点的平行线在等边三角形两边的截距代表该点在对应顶点分组的占比,因此可以得出该点的在三个分组中的占比情况。如图2:过绿色点分别向三条边做平行线,然后按逆时针方向依次读取平行线在三条边的截距约为(0.33, 0.33, 0.33),因此该点在x, y, z 三个组分中的占比为0.33, 0.33, 0.33;依此原则可得出,红点占比为0.1,0.2,0.7;蓝点占比为0.2,0.7,0.1;该结果跟我们的作图代码是相符的。
data <- data.frame( x=c(0.33, 1, 2),
y=c(0.33, 2, 7),
z=c(0.33, 7, 1))
p = ggtern(data, aes(x=x, y=y, z=z)) +
geom_point(size=5, alpha=0.7, color=c("green", "red", 'navy')) +
geom_mask() +
annotate(geom ='text',
x =c(0.33, 1, 2),
y =c(0.33, 2, 7),
z =c(0.33, 7, 1),
vjust=c(1.5, 1.5, 1.5),
angle=c(0, 30, 60),
label=c("(0.33, 0.33, 0.33)", "(0.1, 0.2, 0.7)", "(0.2, 0.7, 0.1)" ),
color=c("green", "red", 'navy')) +
theme_rgbw() + geom_crosshair_tern(size=1)
pggsave(paste0("b2.TenaryPlot.png"), p, width=89, height=89, units="mm")
ggsave(paste0("b2.TenaryPlot.pdf"), p, width=89, height=89, units="mm")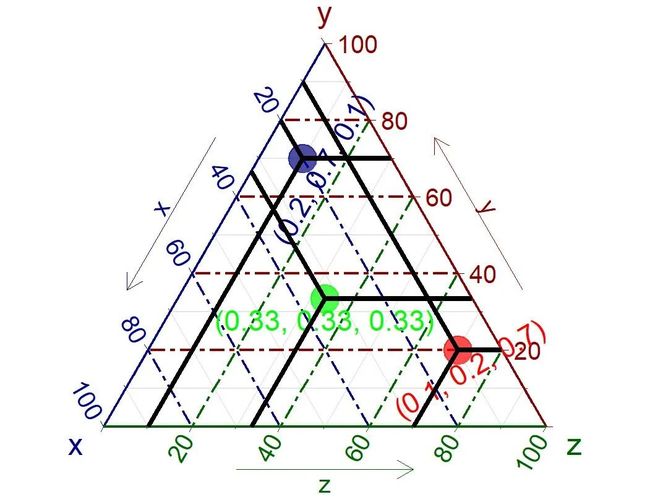
图2. 各点在x, y, z中的占比
在微生物多样性实际分析中,由于OUTs数目繁多,我们并不需要获得如此精确的占比情况。一般来说,三元图不同的点代表不同的OTUs(或其他分类水平),点的大小代表该OTUs的平均丰度(一般需要log2或log10等转换)。不仅如此,还可以对OTUs在各微环境中的丰度数据进行统计检验后,得出各OTU分别在哪种微环境中显著富集,此时三元图不仅表现出OTUs或者物种在微环境中的相对比例,还包含显著性统计结果,它打破了火山图或韦恩图两两比较的结果,总共展示了6次两两比较的结果,即每个组的富集情况是相对于其他两组的。
实例解读
三元图的应用有非常大的局限性,就是必须有3个实验组。只有两组的课题就不要考虑此类图型的应用了。值得一提的是,实验设计3组也是有着最高性价比和最广泛的应用,因为2组只有1种比较组合,而3组仅成本上升50%,却有3种比较组合,结果的图表量瞬间上升为3倍,性价比极高。而3组以上组合太多,反而不利于作者把故事讲清楚,读者阅读和理解也存在一定困难。因此强烈推荐在尽量可能的情况下,设计为3组起的实验,可以有更多的分析和讨论空间,也可以应用有监督的排序分析、三元图等常用结果样式。下面分别选取来自Frontier in Microbiology、PNAS和Nature Communications杂志中近年的三元图结果进行解读和点评。
例1. 三元图门着色+堆叠柱状图
本例2017年发表于自Frontiers in Microbiology文章的图4,描述了植物从三个北高寒带气候区的共同核心类群中组装特定物种的细菌群落(Kumar et al., 2017)。
图3. 三元图展示OTUs的空间特异性
图片描述:三元图展示不同区域(土体土、根际土、根内)样本的群落结构在门水平上的差异情况,每个圆点代表一个OTU,OTU的大小、颜色和位置分别代表其相对丰度、菌门水平名称和分组情况。
Distribution of OTUs and phyla across different compartments. (A) Ternary plot of all OTUs plotted based on the compartment (Bulk soil, Rhizosphere soil, Endosphere) specificity. Each circle represents one OTU. The size, color and position of each OTU represents it relative abundance, bacterial phyla and affiliation of the OTU with different compartments, respectively.
图注描述注意事项:
总述图表展示的信息:
三元图展示OTUs在三个不同部分的空间特异性;
详细描述图片中各元素代表什么:
每个圆点代表一个OTU,OTU的大小、颜色和位置分别代表其相对丰度、菌门水平名称和分组情况。
门着色突出各门在空间的分布,同时配合堆叠柱状图展示门的相对丰度并添加显著性标记方便描述和下结论。
文中的图片解读:
主要描述了比较突出的情况,如丰度较高、规律性变化或明显聚集在某区域的门。
在门水平的不同部位中,细菌群落结构明显不同。这些差异的主要原因是由于厚壁菌门在根内相对丰度较高引起,而在土体土和根际土中它们的丰度却很低。变形杆菌和拟杆菌的相对丰度从土体土到根际土再到根内逐渐增加,同时候选门AD3、芽单胞菌门和绿弯菌门的相对减少,它们共同构成根内微生物群落丰度<4%。
Bacterial community structures were clearly different in the different compartments at the phylum level. These differences were mainly driven by strong relative enrichment of Firmicutes in the endosphere-derived sequence data sets, compared to their very low abundances in the bulk and rhizosphere soils. The relative abundances of Proteobacteria and Bacteroidetes increased progressively from bulk to rhizosphere soil to the endosphere, with a concomitant decrease in those of candidate division AD3, Gemmatimonadetes and Chloroflexi, which collectively constituted <4% of endosphere communities.
例2. 三元图展示各种特异+箱线图
本例选自2016年发表于PNAS杂志上一文的图4(Zgadzaj et al., 2016),介绍了豆科模式植物百脉根的结瘤突变体中根际微生物组变化的研究。
图4. 三元图展示不同取样部位中特异富集的OTUs
图片描述:
最上方的”Wild-type”和”Mutants”指明材料类型,分别为野生型和突变体;
三个顶点分别为三个取样部分,并在括号中指出显著富集的OTUs数量;
点的大小代表三组样品的平均相对丰度;
通过颜色指示显著富集情况:
土壤=土色, 根际土=橘黄色,根系=绿色,灰色=两两比较中未全部显著富集的(均未富集,只相对其中一个分组富集)。
配和箱线图,进一步突出各组特异OTUs累计丰度的组间差异变化。
Ternary plots depicting compartment RA of all OTUs (>5 ‰) for WT SampleID (A; WT; n=73) and mutant SampleID (B; nfr5-2, nfr5-3, nin-2, and lhk1-1; n=118) across three soil batches (CAS8–CAS10). Each point corresponds to an OTU. Its position represents its RA with respect to each compartment, and its size represents the average across all three compartments. Colored circles represent OTUs enriched in one compartment compared with the others (green in root, orange in rhizosphere, and brown in root SampleID). Aggregated RAs of each group of enriched OTUs (root-, rhizosphere- and soil-enriched OTUs) in each compartment for the WT SampleID (C; WT; n=73) and mutant SampleID (D; nfr5-2, nfr5-3, nin-2, lhk1-1; n=118) are shown. In each compartment, the difference from 100% RA is explained by OTUs that are not significantly enriched in a specific compartment.
总结:
该图在例1的基础上添加了组间显著性差异比较的结果,包含了6次两两比较和三次韦恩图比较的结果,信息高度概括。同时,作者还进一步结合相对丰度的箱线图突出组间差异;此外,通过使用相同的色系进行A与B图,C与D图的横向比较,突出明显的分布差异,使用两类材料微生物组不同的规律一目了然。
例3. 三元图分区+堆叠柱状图
本文由荷兰皇家科学院生态研究所的S. Emilia Hannula和中科院遗传发育所朱峰研究员等于2019年8月发表于Nature Communications (https://doi.org/10.1038/s41467-019-09284-w)。揭示了食叶昆虫微生物群落来源于土壤而不是取食植物。中文解读详见:[Nature子刊:植食昆虫微生物组来自土壤](https://mp.weixin.qq.com/s/uiXqcGZEt3QX-V49r88J2w)
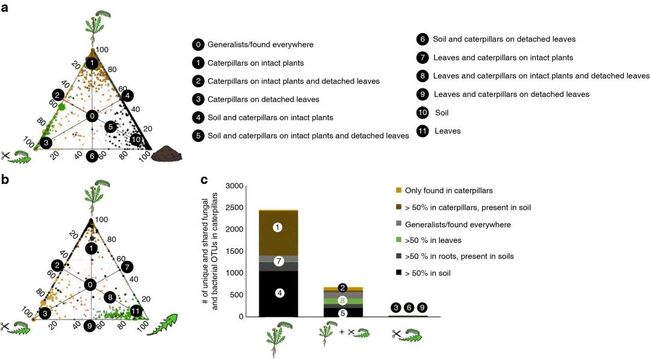
图5. 土壤、叶片和毛虫共享的微生物。
图片描述:
a. 在土壤,食用离体叶片,食用植株的毛虫中共有OTU的分布。
b. 在植物,食用离体叶片,食用植株的毛虫中共有OTU的分布。
c. 食用离体叶片毛虫和植株毛虫肠道共有和特有OTU的数量。图中颜色含义和a,b图中的一致。
a,b 三元图中符号代表一个OTU,细菌使用圆形表示,真菌使用三角形表示。使用OTU为在每组样品中出现频率超过10%的OTU。符号的大小代表了每个OTU的平均相对丰度,颜色代表该OTU主要的来源区域。绿色代表该OTU超过50%来源于叶片,棕色代表该OTU丰度超过50%来自毛虫肠道(在离体叶片上的毛虫颜色为浅棕色,在植株上生长的毛虫颜色为深棕色)。黑色代表OTU在土壤中丰度超过50%,灰色代表OTU在根系中丰度超过50%。灰色的符号代表在全部区域都存在的OTU。每个符号的位置代表该OTU出现在不同样品中的丰度比例。50%的线编号为0-9,详见图例说明具体的分区。
Fig. 2 Bacterial and fungal OTUs shared among caterpillars, plants and soil. a, b Ternary plots of OTUs found in caterpillars. Each symbol represents a single OTU; circles represent bacterial OTUs and triangles fungal OTUs. Only OTUs found in at least 10% of the samples are included in the figure. The size of each symbol represents its relative abundance (weighted average) and its color the compartment where it is primary found. Green depicts OTUs found >50% in leaves, brown depicts OTUs found >50% in caterpillars (dark brown OTUs in caterpillars on intact plants and light brown on detached leaves), black depicts OTUs found >50% in soil, grey OTUs found >50% in roots. Grey symbols represent general OTUs found in all compartments. The position of each symbol represents the contribution of the indicated compartments to the total relative abundance. The 50% lines are drawn in the figure and most important compartments are marked with numbers (0–9). a Depicts OTUs shared between soil (right side), caterpillars on intact plants (top) and caterpillars on detached leaves (left) and b depicts OTUs shared between plants (right), caterpillars on intact plants (top) and caterpillars on detached leaves (left). c The total number of unique and shared OTUs of caterpillars on intact plants and caterpillars on detached leaves. Both fungi and bacteria are included in the figure and their identity on the phylum/class level. The color of the compartment where the OTUs are predominantly found and the corresponding region in panel a and b is also shown.
结果
两种喂养方式毛虫共有的核心微生物群落也存在于植株根系(19.1%)和叶片(20.3%)中 (图2a–c),同时也有特有的微生物群,大约16.7%微生物仅仅发现在毛虫中。毛虫核心微生物群组成细菌主要有变形菌门、放线菌门和厚壁菌门,真菌为未注释的OTU。值得注意的是以完整植株为食的毛虫大部分OTU也存在与土壤中(75%)。以完整植株为食的毛虫中有超过离体叶片为食的毛虫三倍的OTU数量。
Caterpillars fed on intact plants and detached leaves shared a common core microbiome which was also present in the leaves (20.3% of their microbiome) and in the roots (19.1%) (Fig. 2a–c), but also harbored unique microbes; 16.7% of the caterpillar microbiome was found only in caterpillars. This core microbiome of caterpillars consisted predominantly of Proteobacteria, Acidobacteria, Firmicutes, and unclassified fungi (Supplementary Figs 6, 7). Remarkably, for caterpillars fed on intact plants, a large proportion of the OTUs found in caterpillars, was also detected in the soil (75%; represented as numbers 1 and 4 in Fig. 2a). Microbiomes of caterpillars fed detached leaves had virtually no additional OTUs that were not also found in caterpillars kept on intact plants (Fig. 2c), but the microbiomes of the latter contained three times more OTUs.
总结
本示例对三元图进一步划分区域,并标注编号,自定义定义各区域的分组和特点,是一种非常好的讨论方式;
各区域的顶点采用模式图的方式展示各组,可以有效的提高图片的美观和吸引力;
四组时,选择想要表达的3组,可以展示两个2-4个三元图,本文两个三元图分两次讨论,进一步增加结果的丰富性和复杂性;
结合柱状图的比较,还有之前的箱线图,都是递进表达的方式,让读者更容易理解复杂的问题,同时保持图片兼顾颜值和可读性。
绘图实战
本项目的文件代码、相关R包可通过github、公网地址和百度网盘3种方式下载。关于更多本项目中示例文件的下载,R包安装的内容,请参考之前的章节:
211.Alpha多样性箱线图(样章,11图2视频)
由于前期数据处理是三元图主要的难点,所以在这里将数据处理和可视化分开,使用时便于检查异常、调整分析细节。
环境设置
本教程需要在R语言环境下运行,推荐在RStudio界面中学习。目前测试版本为:Windows 10,R 4.0.x和 RStudio 1.3.x。理论上Mac、Linux系统,以及R或RStudio的新版本是兼容的,但并没有广泛测试,有问题尝试搜索自行解决、欢迎分享经验。
按需求安装,没必要每次都运行该安装代码,一般运行一次即可。
p_list = c("tidyverse", "ggtern", "BiocManager")
for(p in p_list){
if (!requireNamespace(p, quietly = TRUE))
install.packages(p)}
if (!requireNamespace("edgeR", quietly=T))
BiocManager::install("edgeR")数据处理
OTU表数据源来自MicrobiomeStatPlot项目中Data/Science2019目录中的otutab.txt(https://github.com/YongxinLiu/MicrobiomeStatPlot/blob/master/Data/Science2019/otutab.txt)。
函数data_clean参数介绍:
输入:
特征表和样本元数据
输出:
各组均值,用于三元图中的位置
otu: 特征表,如OTU/ASV表
design: 实验设计信息,样品信息的列名设置为SampleID,分组信息列名设置为Group
type: otu_table 类型,绝对丰度absolute/相对丰度relative;
如果是绝对丰度,一定要注明。
threshold: 相对丰度阈值,希望丢弃(获取)的相对丰度大小
times: 点的倍数变化,主要跟可视化有关,如果结果图中的legend全是小数,可以通过倍数变化将其转变为正数,也可以在可视化时自己调整,不影响其传递的丰度相对大小信息。
library(tidyverse)
# 数据处理函数
data_clean <- function(otu, design, type=c("relative", "absolute"), threshold=0.001, times=100){
# 函数测试数据
# library(amplicon)
# otu=otutab
# metadata$SampleID=rownames(metadata)
# design=metadata[,c("SampleID","Group")]
# type="absolute"
# threshold=0.0005
# times=100
# 绝对丰度转相对丰度1
if (type == "absolute"){
otu_relative <- apply(otu, 2, function(x){x/sum(x)})
}else {otu_relative <- otu}
# 至少有一个样本大于阈值即保留
idx <- rowSums(otu_relative > threshold) >= 1
otu_threshold <- as.data.frame(otu_relative[idx, ])
otu_threshold$OTUs <- row.names(otu_threshold)
#转换宽表格为长表格
otu_longer <- pivot_longer(data=otu_threshold,
cols=-OTUs,
names_to="SampleID",
values_to="value")
# 按"SampleID"对应添加元数据中的分组Group
merge_data <- merge(otu_longer, design, by ="SampleID")
# 去除样本列
# otu <- subset(merge_data, select=-SampleID)
# 元数据不只有样本列,直接筛选OTUs、Group和value更稳健
otu <- subset(merge_data, select=c("Group","OTUs","value"))
# 按OTUs和Group求均值
otu_mean <- otu %>% group_by(OTUs, Group) %>%
summarise(value=mean(value))
# 转换回宽表格
otu_tern <- otu_mean %>%
group_by(Group, OTUs) %>%
mutate(index=row_number()) %>%
pivot_wider(names_from=Group,values_from=value) %>%
select(-index)
# 此处用group_by可以直接合并均值,不用长、宽转换两次
# 调整点大小均值,可缩放 size of points
otu_tern$size <- (apply(otu_tern[2:4], 1, mean))*times
return(otu_tern)
}# 读取输入文件
otutab <- read.delim("otutab.txt", header=T, row.names=1)
design <- read.delim("metadata.txt", header=T, row.names=NULL)
# 只提取元数据中的样本名和分组列,要求名称为SampleID和Group
design = design[,c("SampleID","Group")]
# 计算三元图输入数据:各组相对丰度均值
otu_tern <- data_clean(otutab, design, type="absolute", threshold=0.001, times=100)
head(otu_tern,n=3)ggtern可视化
ggtern 是Nicholas Hamilton开发的,用于创建三元图的ggplot2的扩展包,详细参数和用法见?ggtern或官方说明文档。
library(ggtern)
# x/y/x指定三个组名,显示顺序为左、中、右
p <- ggtern(data=otu_tern, aes(x=KO, y=OE, z=WT)) +
geom_point(aes(size=size), alpha=0.8, show.legend=T) +
scale_size(range=c(0, 6)) + geom_mask() +
guides(colour="none") + theme_bw() +
theme(axis.text=element_blank(), axis.ticks=element_blank())
pggsave(paste0("p1.png"), p, width=89, height=59, units="mm")
ggsave(paste0("p1.pdf"), p, width=89, height=59, units="mm")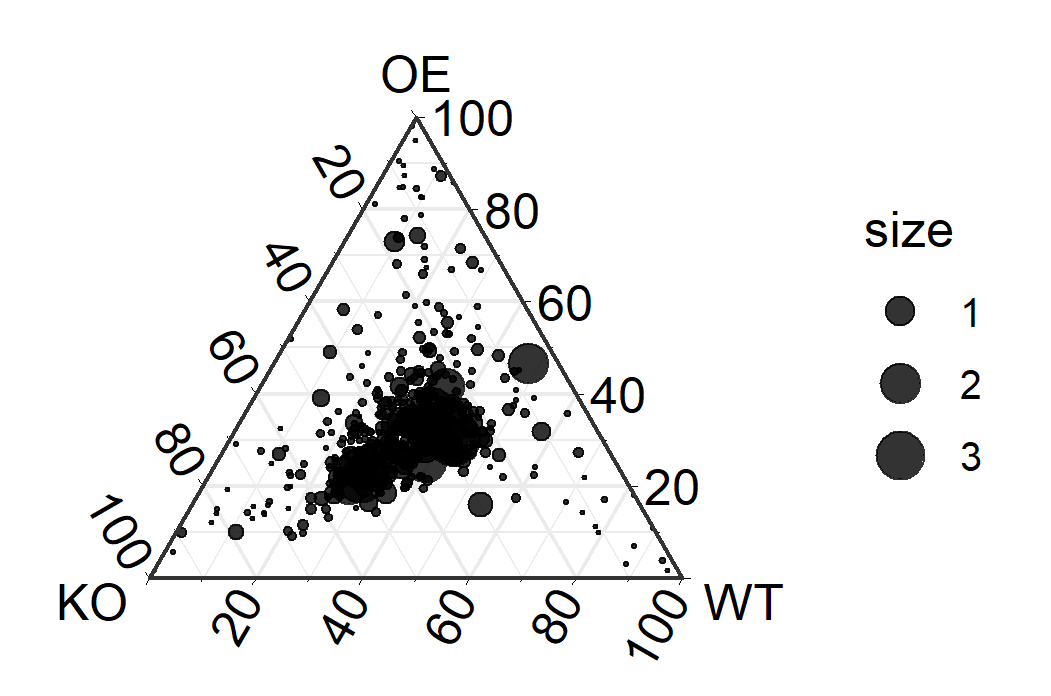
图6. 三元图展示OTUs的实验分组特异性
图片描述:
三元图展示不同处理组(KO, OE, WT)的样本的群落结构在OTUs上的丰度差异情况,每个圆点代表一个OTU,OTU的大小和位置分别代表其相对丰度和分组情况。
图注描述注意事项:
由于该图中并未全部展示所有OTUs,所以在描述的时候,最好对数据筛选的参数进行描述:
图中只展示了相对丰度>0.1%的OTUs;
点大小对应的平均相对丰度的倍数变化;
从图中可以看到KO(基因敲除,knock-out)组与OE(过表达,over-expression)和WT(野生型,wild-type)组存在丰度差异,即基因的有无可对微生物群落的丰富度引起变化。
高丰度特征着色
上图能够展示的信息和按主丰度着色后的可视化方案是一致的,但叙述时可以像图3一样着重介绍突出情况,比如丰度较高的OTUs有那些,富集情况很特殊的有哪些。
函数top_OTUs参数介绍:ggtern输入文件筛选丰度前N(10)的OTUs
data: ggtern输入文件
rank: 希望进行着色的丰度排名,推荐10个。
如果超过10个,则需要自己制定配色方案。
top_OTUs <- function(data, rank=10){
# 按丰度size降序排列
data_order <- data[order(data$size, decreasing=T), ]
# if (missing(rank))
# rank=10
# 提取前N行
top <- data_order[1:rank, ]
# 其余部分
otu_el <- data_order[-(1:rank), ]
# 返回top和其它的列表
return(list(top, otu_el))
}plot_data <- top_OTUs(otu_tern, rank=10)
# 配色方案
platte <- c('#a6cee3', '#1f78b4', '#b2df8a', '#33a02c', '#fb9a99',
'#e31a1c', '#fdbf6f', '#ff7f00', '#cab2d6', '#6a3d9a')
p <- ggtern(data=otu_tern,
aes(x=KO, y=OE, z=WT)) +
geom_mask() +
geom_point(data=plot_data[[2]], aes(size=size), color="grey",
alpha=0.8, show.legend=F) +
geom_point(data=plot_data[[1]], aes(size=size, color=OTUs),
show.legend=T) +
scale_colour_manual(values=platte) +
scale_size(range=c(0, 6)) +
# legend
guides(size="none") +
theme_bw() +
theme(axis.text=element_blank(),
axis.ticks=element_blank())
pggsave(paste0("p2.png"), p, width=89, height=59, units="mm")
ggsave(paste0("p2.pdf"), p, width=89, height=59, units="mm")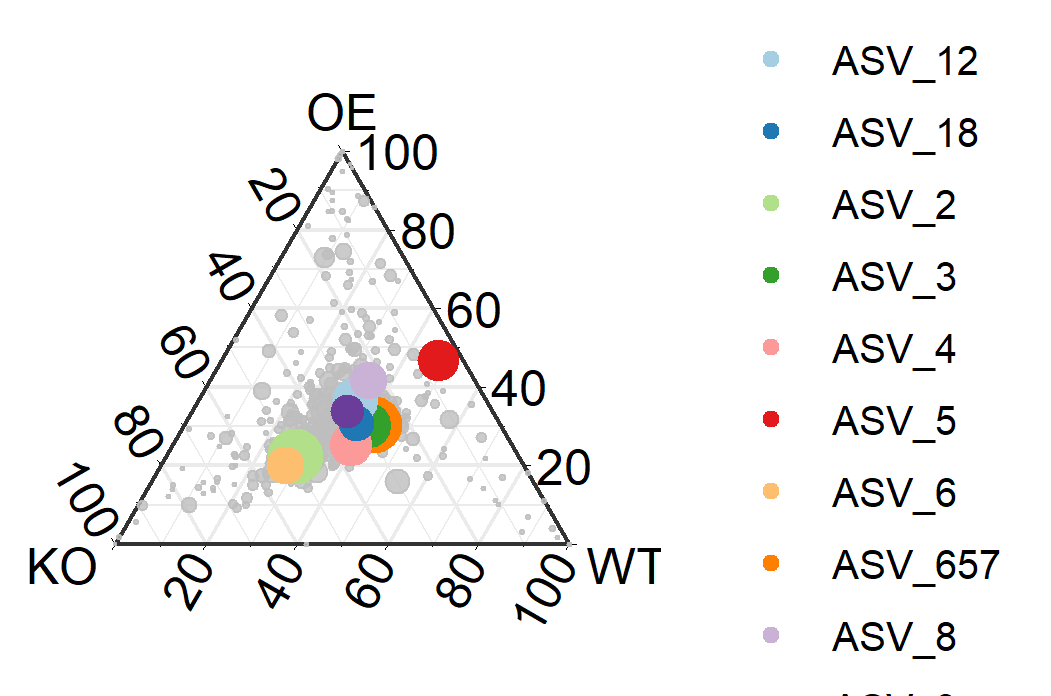
图7. 三元图展示OTUs的实验分组特异性
图片描述:
三元图展示不同处理组(KO, OE, WT)的样本的群落结构在OTUs上的丰度差异情况,每个圆点代表一个OTU,OTU的大小、颜色和位置分别代表其相对丰度、主丰度OTUs名称和分组情况。
值得注意的是,红色原点所代表的ASV_5在KO组中丰度几乎为0,但在WT和OE均有相对较高的丰度。具体可以再结合物种注释进一步描述。
富集显著性分析
除了上面直接展示OTUs在不同分组中的丰度富集情况,还可以通过显著性富集分析,获得在不同分组中显著富集的OTUs,最后进行可视化(例2)。
library(edgeR)
# 计算3组比较各组特有的特征
enrich_data <- function(otu, design, p.value=0.05, adjust.method="fdr"){
# 函数测试数据
# library(amplicon)
# otu=otutab
# metadata$SampleID=rownames(metadata)
# design=metadata[,c("SampleID","Group")]
# p.value=0.05
# adjust.method="fdr"
dge_list <- DGEList(counts=otu, group=design$Group)
# Remove the lower abundance/(cpm, rpkm)
keep <- rowSums(dge_list$counts) >= 0
dge_keep <- dge_list[keep, ,keep.lib.sizes=F]
# scale the raw library sizes dgelist
dge <- calcNormFactors(dge_keep)
# fit the GLM
design.mat <- model.matrix(~ 0 + dge$samples$group)
d2 <- estimateGLMCommonDisp(dge, design.mat)
d2 <- estimateGLMTagwiseDisp(d2, design.mat)
fit <- glmFit(d2, design.mat)
#######
# if (missing(adjust.method))
# adjust.method="fdr"
# if (missing(p.value))
# p.value=0.05
group_index <- as.character(design$Group[!duplicated(design$Group)])
# enrich groups
lrt_1_2 <- glmLRT(fit, contrast=c(1, -1, 0))
lrt_1_3 <- glmLRT(fit, contrast=c(1, 0, -1))
de_1_2 <- decideTestsDGE(lrt_1_2, adjust.method=adjust.method,
p.value=p.value)
de_1_3 <- decideTestsDGE(lrt_1_3, adjust.method=adjust.method,
p.value=p.value)
rich_1 <- rownames(otu)[de_1_2 == 1 & de_1_3 == 1]
enrich_1 <- data.frame(OTUs=rich_1,
enrich=rep(group_index[1], length(rich_1)))
###############################
lrt_2_3 <- glmLRT(fit, contrast=c(0, 1, -1))
lrt_2_1 <- glmLRT(fit, contrast=c(-1, 1, 0))
de_2_3 <- decideTestsDGE(lrt_2_3, adjust.method=adjust.method,
p.value=p.value)
de_2_1 <- decideTestsDGE(lrt_2_1, adjust.method=adjust.method,
p.value=p.value)
rich_2 <- rownames(otu)[de_2_3 == 1 & de_2_1 == 1]
enrich_2 <- data.frame(OTUs=rich_2,
enrich=rep(group_index[2], length(rich_2)))
###################
lrt_3_1 <- glmLRT(fit, contrast=c(-1, 0, 1))
lrt_3_2 <- glmLRT(fit, contrast=c(0, -1, 1))
de_3_1 <- decideTestsDGE(lrt_3_1, adjust.method=adjust.method,
p.value=p.value)
de_3_2 <- decideTestsDGE(lrt_3_2, adjust.method=adjust.method,
p.value=p.value)
rich_3 <- rownames(otu)[de_3_1 == 1 & de_3_2 == 1]
enrich_3 <- data.frame(OTUs=rich_3,
enrich=rep(group_index[3], length(rich_3)))
enrich_index <- rbind(enrich_1, enrich_2, enrich_3)
return(enrich_index)
}enrich_index <- enrich_data(otutab, design, p.value=0.05)
plot_data <- merge(otu_tern, enrich_index, by="OTUs", all.x=T)
p <- ggtern(data=plot_data,
aes(x=KO, y=OE, z=WT)) +
geom_mask() + # 可将超出边界的点正常显示出来
geom_point(aes(size=size, color=enrich),alpha=0.8) +
guides(size="none") +theme_bw() +
theme(axis.text=element_blank(),
axis.ticks=element_blank())
pggsave(paste0("p3.png"), p, width=89, height=59, units="mm")
ggsave(paste0("p3.pdf"), p, width=89, height=59, units="mm")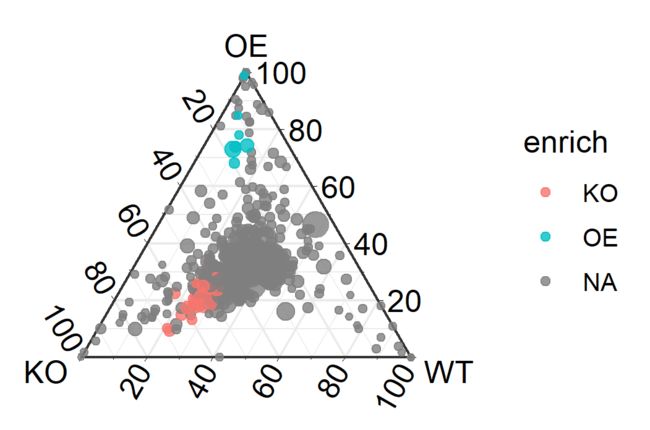
图8. 三元图展示各组特异富集的OTUs
图片描述:
除了图6中的信息外,该图还展示了在KO, OE, WT三个分组中,分别相对于另外两个分组显著性富集的OTUs。WT组一个特异的都没有。
图注描述注意事项:
由于该图中并未全部展示所有OTUs,所以在描述的时候,最好对数据统计分析过程的参数进行描述:
相对丰度阈值,如只展示对丰度>0.1%的OTU/ASV
点大小对应的平均相对丰度的倍数变化,因为相对丰度值太小,这里点大小放大了一百倍;
组间比对时要定义统计方法和p.value和adjust.method,如这里使用edgeR包的glmLRT差异比较方法,P值<0.05,校正方法为FDR。
参考文献
Wikipedia. Ternary_plot. https://en.wikipedia.org/wiki/Ternary_plot
Manoj Kumar, Günter Brader, Angela Sessitsch, Anita Mäki, Jan D. van Elsas & Riitta Nissinen. (2017). Plants Assemble Species Specific Bacterial Communities from Common Core Taxa in Three Arcto-Alpine Climate Zones. Frontiers in Microbiology 8, doi: https://doi.org/10.3389/fmicb.2017.00012
Rafal Zgadzaj, Ruben Garrido-Oter, Dorthe Bodker Jensen, Anna Koprivova, Paul Schulze-Lefert & Simona Radutoiu. (2016). Root nodule symbiosis in Lotus japonicus drives the establishment of distinctive rhizosphere, root, and nodule bacterial communities. Proceedings of the National Academy of Sciences of the United States of America 113, E7996-E8005, doi: https://doi.org/10.1073/pnas.1616564113
S. Emilia Hannula, Feng Zhu, Robin Heinen & T. Martijn Bezemer. (2019). Foliar-feeding insects acquire microbiomes from the soil rather than the host plant. Nature Communications 10, 1254, doi: https://doi.org/10.1038/s41467-019-09284-w
刘永鑫,扩增子图片解读7三元图:美的不要不要的,再多用也不过分,宏基因组,https://mp.weixin.qq.com/s/R0U_sZlay3gDu5uH1ABYBA
责编:刘永鑫 中科院遗传发育所
版本更新历史
1.0.0,2020/6/6,吴一磊,初稿;文涛审阅;刘永鑫审阅
1.0.1,2020/6/9,吴一磊,小修,添加模拟数据,可独立运行的代码;刘永鑫审阅
1.0.2,2020/6/22,吴一磊,大修,添加实例讲解,代码封闭函数;刘永鑫审阅
1.0.3,2020/6/25,席娇,文字修改
1.0.4,2020/6/27,刘永鑫,整合修改意见;添加一篇NC实例讲解
1.0.5,2020/6/28,刘永鑫,终审、格式调整、排版
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读