DeepMind开源AlphaFold,蛋白质预测模型登上《Nature》
| 2020-01-19 16:09 |
导语:CNN+Rosetta

雷锋网(公众号:雷锋网)报道:还记得AlphaFold成名的那一战么?
2018年的11月2日,在第13届全球蛋白质结构预测竞赛(CASP)上,AlphaFold获得了预测43种蛋白中的25种蛋白结构的最高分,在98名参赛者中排名第一。对于DeepMind的预测方法,由于当时没有具体论文发布,众多学者认为是计算能力突出使得AlphaFold获得冠军。

1月15日,DeepMind关于AlphaFold模型与代码通过了同行评审发布在了杂志《nature》上面。并且模型和代码已经开源。
代码:https://github.com/deepmind/deepmind-research/tree/master/alphafold_casp13
模型:https://www.biorxiv.org/content/10.1101/846279v1.full.pdf
根据DeepMind的介绍,在预测蛋白质结构的物理性质方面使用了两种不同的方法来构建预测模型。并且两种方法都是基于深度神经网络来设计的,另外,预测模型主要完成对基因序列中蛋白质的特性的预测,这些特性包括:a:成对的氨基酸之间的距离;b:连接这些氨基酸的化学键之间的角度。

两种图片展示方式都表达了AlphaFold预测的准确度,像素亮度代表氨基酸之间的距离,像素颜色越亮,两个残基(residues)对就越近。第一行图片是真实距离,中间一行图片展示的是平均距离。
具体操作步骤为用神经网络预测蛋白质中的每一对残基的概率分布,然后将这些概率合并为一个分数,从而能够估计预测蛋白质结构的准确性。另外,还训练了一个单独的神经网络,基于所有距离的总和来评估预测的蛋白质结构与实际的结构的接近程度。然后使用评分系统(也是用神经网络构建)找到最优的预测。

雷锋网:整个蛋白质结构预测的过程
第二种方法是采用梯度下降优化分数,从而达到更准确的精度。更具体的是将梯度下降应用在整个蛋白质结构链的预测过程,而不仅仅是蛋白质展开的“碎片”。
模型简介

https://www.biorxiv.org/content/10.1101/846279v1.full.pdf
根据DeepMind公开的论文《Improved protein structure prediction using predicted inter-residue orientations》。AlphaFold由深度学习和传统算法混合而成:CNN+Rosetta。
具体来说构建神经网络预测残基间的方向和距离,然后用Rosetta对蛋白质结构同源建模、结构修复。同时设计一种Rosetta的优化方法来补充Rosetta能量函数的预测约束,进而生成更精确的模型。
尽管训练数据集全部为天然的蛋白质,但该模型始终将更高的概率分配给新设计的蛋白质,并找到决定残基的关键折叠和建立蛋白质结构“理想性(ideality)”的独立定量衡量标准。
更为具体的,模型的由两个关键部分组成:一个是深度残差卷积神经网络,即将多序列比对( multiple sequence alignments)作为输入;输出的信息为蛋白质中的残基对中的相对距离和方向。
另一个是在网络输出的基础上针对残基对的距离和方向最小化的约束建立快速Rosetta模型(fast Rosetta model)在训练数据集方面,DeepMind使用的蛋白质数据库(PDB)里面有15051个蛋白质链条信息,其中有30%是被标注的数据。
关于对模型的测试,DeepMind使用的是两个独立的测试集:第一个来自CASP13,第二个来自CAMEO实验。另外,在CASP13数据集上,DeepMind使用完整的蛋白质序列而不是结构域序列来模拟。
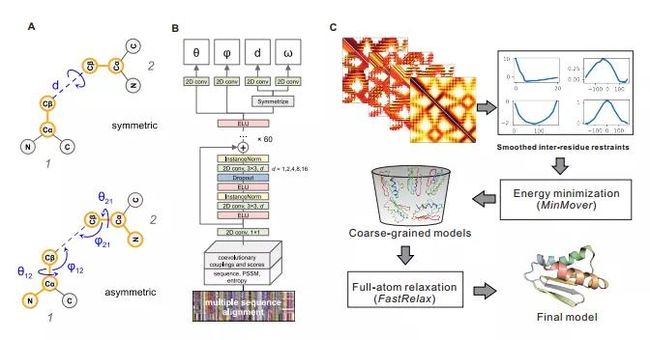
图注:A:用角度和距离表示从一个残基到另一残基的转换。B:神经网络结构根据MSA预测残基间的几何形状;C:预测过程概览
在残基间的几何预测方面,DeepMind使用深度残差神经网络。与大多数预测方法不同,DeepMind除了预测化学键之间的距离之外,残基对之间的方位也属于其预测的范围。如上图所示,残基1与残基2之间的方位由3个二面角以及2个平面角表示。其中ω表示沿虚轴(连接两个残基的原子)旋转角度。平面角定义了从残基1看到残基2的Cβ原子的方向。另外,与d和ω不同,θ和φ坐标是不对称的,其取决于残基的顺序。综上6个参数d,ω,![]()
![]() 定义了两个残基的主干原子的相对位置,这6个参数也是神经网络所要预测的。
定义了两个残基的主干原子的相对位置,这6个参数也是神经网络所要预测的。
神经网络的输入是从MSA提取的特征,并且执行动态运算。输入的一维特征包括:蛋白质氨基酸序列的独热编码、位置特定频率矩阵(position-specific frequency matrix)、配位熵(positional entropy)。随后这些一维特征被水平和垂直平铺,然后堆叠在一起以产生2×42=84个2维特征映射。此外,DeepMind从MSA中提取配对统计信息,这些信息从输入MSA构造的缩小协方差矩阵的逆导出的耦合来表示。首先计算一个站点(one-site)和两个站点(two-site)的频率计数,计算公式如下:

其中,A和B分别表示氨基酸的属性,δ是克罗内克函数,Wm是MSA中序列数目的倒数,与序列m至少有80%的序列同源性。其中

然后计算样本协方差矩阵,公式如下:

接下来在收缩(即通过在对角线上附加恒定权重进行正则化)之后求其逆(也称为精度矩阵):

范数转换:
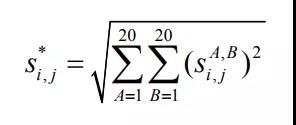
最后修正:

在具体的训练过程中,DeepMind使用交叉熵进行评估,其评估结果是总损失等于4个拥有同样权重的分支网络的损失之和。另外,使用学习率为1e-4的Adam优化器,Dropout保持85%的概率。并随机地对输入MSAs在对数尺度上平均进行采样,长度超过300个氨基酸的大蛋白质会被随机切片。然后对于距离分布,将概率值通过以下等式将概率值转换为分数:

上述公式中的a是常数(=1.57),di是第i个bin的距离,
pi是第i个bin的距离的概率,N是bin的总数。
对于方向的分布,不考虑归一化的情况下,概率转分数的公式为:
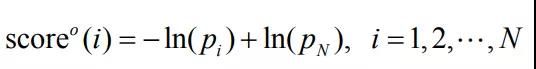
最后将所有的分数通过Rosetta转化成平滑势能,并用约束限制势能最小化。综上整个模型我们可以归结为:扔进一个蛋白质序列,生成一个结构、一个评分,通过评分判断是否满意,不满意就再生成一个。
代码简介
根据DeepMind提供的Github中介绍,这份代码包括预测网络、相关的模型权重以及在《自然》杂志上发表的CASP13数据集的实现。此外,还详细的介绍了数据的下载地址,以及如何输入数据;运行系统如何操作也给出了详细的指导。雷锋网 AI科技评论在下方再次给出github地址,感兴趣的读者赶紧去上手试一试吧!
https://github.com/deepmind/deepmind-research/tree/master/alphafold_casp13