1. 资料下载
仅下载CEL.gz结尾的八个文件
https://uon.ent.box.com/s/iyy15wqlwcsuugrbe6kxlxcf3ztmztw8/folder/32707318961
- 解压后生成八个文件
-
该实验有1个对照和3个处理,各有2个重复,共8张芯片
 image.png
image.png
2. 读取CEL文件
- 安装两个包
>source("http://bioconductor.org/biocLite.R")
>biocLite(c("affy","simpleaffy"))
# 设置工作路径
setwd("/Users/chengkai/Desktop/PMRA/CEL")
library(affy)
# 建立文件过滤器
filters<-matrix(c("CEL file",".[Cc][Ee][Ll]","All",".*"),ncol=2,byrow= T)
# 使用tk_choose.files函数选择文件
cel.files<-tk_choose.files(caption="Select CELs",multi=TRUE,filters= filters,index=1)
# 注意:较老版本的tk函数有bug,列表的第一个文件名可能是错的
basename(cel.files)

image.png
# 用ReadAffy( )函数读如CEL文件
data.raw<-ReadAffy(filenames= cel.files)
sampleNames(data.raw)
old.names <- sampleNames(data.raw)
sampleNames(data.raw) <- paste("CHIP",1:length(cel.files),sep="-") #改名
sampleNames(data.raw) #查看新名称
image.png
3. 查看芯片的基本信息
pm.data <- pm(data.raw)
head(pm.data) # Perfect-match probes
mm.data <- mm(data.raw)
head(mm.data) # Mis-match probes
head(geneNames(data.raw)) # ProbeSet names
sampleNames(data.raw) # Sample names
(pdat <- pData(data.raw)) # Phenotypic data

image.png
4. 显示芯片扫描图像(灰度)
n.cel <- length(cel.files) #芯片数量
par(mfrow=c(ceiling(n.cel/2),2))
par(mar=c(0.2,0.2,2,0.2))
pallette.gray <- c(rep(gray(0:10/10), times = seq(1,41,by=4))) #设置调色板颜色为灰度
for(i in 1:n.cel) image(data.raw[,i], col=pallette.gray) #通过for循环逐个作图
- 如果图像特别黑,说明型号强度低;如果图像特别亮,说明信号强度很有可能过饱和
-
如果芯片图像有斑块现象就很可能是坏片。
image.png
5. 对灰度值做简单统计分析
par(mfrow=c(1,1))
par(mar=c(4,4,3,0.5))
par(cex = 0.7)
if(n.cel>40) par(cex = 0.5)
cols <- rainbow(n.cel*1.2) #rainbow是R的一个函数,用于产生彩虹色
boxplot(the.data, col = cols, xlab="Sample", ylab="Log intensity")

image.png
par(mar = c(4,4,3,0.5))
hist(data.raw, lty=1:3, col = cols)
legend("topright", legend = sampleNames(data.raw),
lty = 1:3, col = cols, box.col = "white", xpd = TRUE)
box()

image.png
6. MA-plot分析
par(mfrow=c(ceiling(n.cel/2),2))
par(mar=c(3,3,2,0.5))
par(tcl=0.2)
par(mgp=c(2,0.5,0))
MAplot(data.raw, cex=0.8)
-
IQR差别大的芯片可能有问题,但芯片能不能用得看具体情况(参考其他指标)而定。
 image.png
image.png
7. RNA降解分析
par(mfrow=c(1,1))
par(mar=c(4,4,3,0.5))
RNAdeg <- AffyRNAdeg(the.data)
summaryAffyRNAdeg(RNAdeg)
plotAffyRNAdeg(RNAdeg, cols = cols)
legend("topleft", legend = sampleNames(the.data), lty=1, col = cols, box.col = "white", xpd = TRUE)
box()
-
理想状况下各样品的线(分段)是平行的。从上面图上看芯片1可能有点问题。
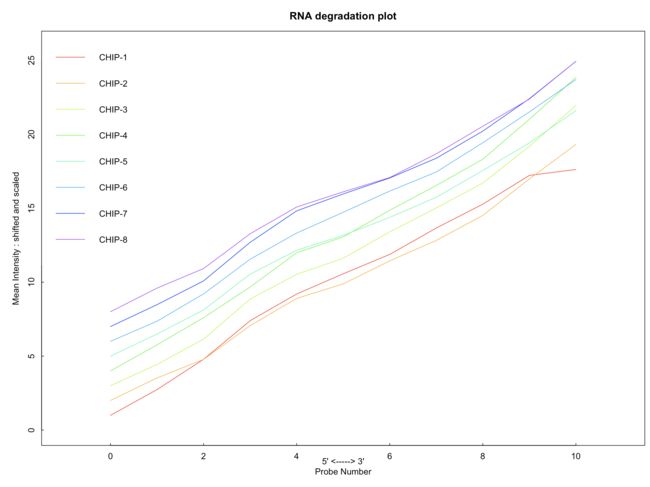 image.png
image.png
8. 用simpleaffy包进行分析
> library(gcrma)
> library(genefilter)
> library(simpleaffy)
> qc.data <- qc(data.raw) #计算芯片质量数据
> (avbg.data <- as.data.frame(sort(avbg(qc.data)))) #平均背景值,如果太大则表示可能有问题
sort(avbg(qc.data))
CHIP-8 60.74498
CHIP-5 63.53240
CHIP-2 63.71067
CHIP-3 63.92073
CHIP-7 63.92203
CHIP-6 66.59299
CHIP-1 78.94698
CHIP-4 79.61115
> (sfs.data <- sort(sfs(qc.data))) #样品的scale factor
[1] 0.5689041 0.6235015 0.6904516 0.6920065 0.7659872 0.8178757 0.8190931 0.8386477
> max(sfs.data)/min(sfs.data) #affy建议每个样品间的sf差异不能超过3倍
[1] 1.474146
> as.data.frame(percent.present(qc.data)) #表达基因所占的比例,太小则表示有问题
percent.present(qc.data)
CHIP-1.present 58.26830
CHIP-2.present 62.09996
CHIP-3.present 62.98115
CHIP-4.present 60.94695
CHIP-5.present 58.01841
CHIP-6.present 59.35116
CHIP-7.present 62.65673
CHIP-8.present 62.29724
> ratios(qc.data) #内参基因的表达比例
actin3/actin5 actin3/actinM gapdh3/gapdh5 gapdh3/gapdhM
CHIP-1 0.3859601 -0.297736190 0.3117987 -0.9426138
CHIP-2 0.3999174 -0.179445980 0.3333365 -0.6740746
CHIP-3 0.3891463 -0.005161427 0.5413926 -0.7286380
CHIP-4 0.4888751 -0.152291418 0.5449479 -0.7080973
CHIP-5 0.2048606 -0.348223137 0.4259705 -0.6383242
CHIP-6 0.4553779 -0.039076158 0.2425867 -0.8056763
CHIP-7 0.5528239 -0.226408224 0.4425869 -0.5121321
CHIP-8 0.4545361 -0.152245701 0.2307999 -0.8547676
参考文献
- http://seuzsl.blog.163.com/blog/static/2187980520134910258605/