title: DALS022-机器学习01-聚类与热图
date: 2019-08-22 12:0:00
type: "tags"
tags:
- 聚类
- PCA
categories: - Data Analysis for the life sciences
前言
这一部分是《Data Analysis for the life sciences》的第9章机器学习的第1小节,这一部分的主要内容涉及聚类与热图,相应的Rmarkdown文档可以参考作者的Github。
机器学习是一个非常广泛的主题和高度活跃的研究领域。 在生命科学中,涉及到“精准医学”的大部分内容都是与机器学习在生物医学数据方面的处理有关。 常规的思路就是从检测的指标中预测或发现一些信息。例如,我们能够从基因表达谱中发现新的癌症吗?我们能通过一系列的基因型来预测药物反应吗?在这一部分中,我们主要介绍两个机器学习的方法,即聚类(clustering)和类预测(class prediction)。
聚类
我们还使用前面的组织基因表达的数据来深圳一下聚类的概念和思路:
library(tissuesGeneExpression)
data(tissuesGeneExpression)
为了说明聚类在生命科学方面的应用,我们先假设我们并不知道上面的几个样本是不同的组织,我们通过聚类来看一下它们的表达谱情况,第一步就是计算不同的样本之间的距离,如下所示:
d <- dist( t(e) )
层次聚类
当我们计算了每对样本之间的距离后,我们需要使用聚类算法将它们聚成不同的组。层次聚类算是众多聚类算法中的一个。每个样本首先会被当作一组(group),然后不断地通过聚类算法迭代,将两个相似的组结合起来,一直到所有的样本都聚为一组。对于样本之间的距离我们已经了解了,但是,不同组之间的距离我们并不了解。关于组与组之间的距离计算方法有很多种,这些方法的核心都是通过计算组与组之间的成员的距离实现的。具体的可以查看hclust()函数的帮助文档。
我们通过hclust()函数来对不同组之间的距离进行层次聚类分析。这个函数会返回一个hclust对象,它描述的我们通过上述算法进行的组划分(grouping)。随后使用plot()函数绘制出树状图,如下所示:
library(rafalib)
mypar()
hc <- hclust(d)
hc
plot(hc,labels=tissue,cex=0.5)
结果如下所示:
> library(rafalib)
> mypar()
> hc <- hclust(d)
> hc
Call:
hclust(d = d)
Cluster method : complete
Distance : euclidean
Number of objects: 189
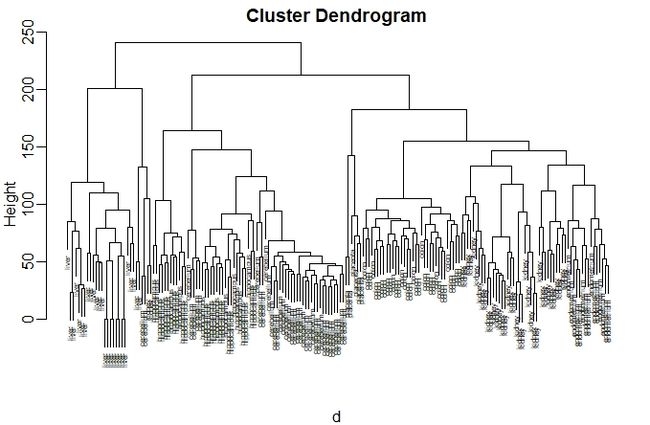
使用这种算法能否发现不同组织的簇(clusters)?在上面的这个图形里,我们不太容易发现这些不同的组织,因此我们需要使用myclust()函数来给它们加上颜色,如下所示:
library(rafalib)
myplclust(hc, labels=tissue, lab.col=as.fumeric(tissue), cex=0.5)
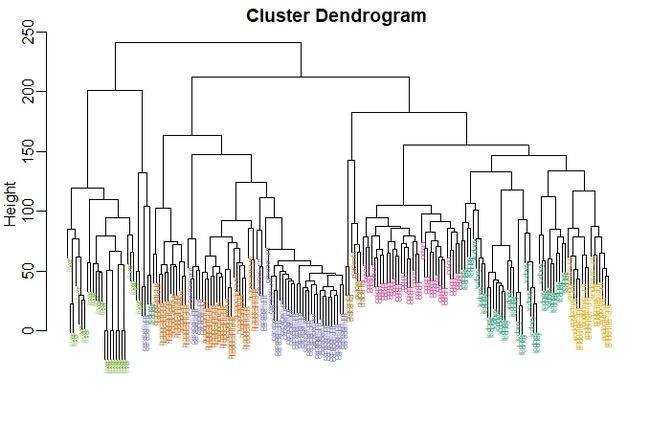
从图形上我们可以看出来,似乎聚类算法能够发现这些不同的组织。但是,层次聚类并没有定义特定的簇,而是定义了上在树状图。从树状图上我们可以描述任意两组之间的距离。为了定义一些簇,我们需要将把树在某些距离上”切开“,所有的样本在这个距离以下分成不同的组。我们可以绘制出一条水平线,我们就使用120这个距离来切,如下所示:
myplclust(hc, labels=tissue, lab.col=as.fumeric(tissue),cex=0.5)
abline(h=120)
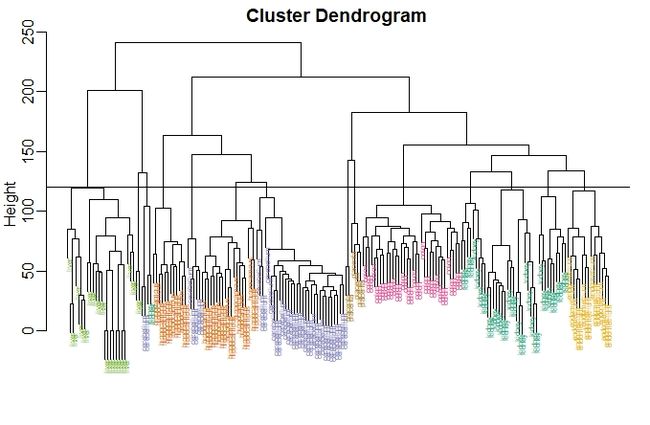
当我们在高度为120上对树进行切割时,我们可以看一下这个线能够把线以下的样本分为几组,如下所示:
hclusters <- cutree(hc, h=120)
table(true=tissue, cluster=hclusters)
计算结果如下所示:
> table(true=tissue, cluster=hclusters)
cluster
true 1 2 3 4 5 6 7 8 9 10 11 12 13 14
cerebellum 0 0 0 0 31 0 0 0 2 0 0 5 0 0
colon 0 0 0 0 0 0 34 0 0 0 0 0 0 0
endometrium 0 0 0 0 0 0 0 0 0 0 15 0 0 0
hippocampus 0 0 12 19 0 0 0 0 0 0 0 0 0 0
kidney 9 18 0 0 0 10 0 0 2 0 0 0 0 0
liver 0 0 0 0 0 0 0 24 0 2 0 0 0 0
placenta 0 0 0 0 0 0 0 0 0 0 0 0 2
从上面可以看出来,分为了8组,另外通过cutreee()函数,我们可以直接指定返回几组簇,这个函数会自动返回结果,如下所示:
hclusters <- cutree(hc, k=8)
table(true=tissue, cluster=hclusters)
结果如下所示:
> hclusters <- cutree(hc, k=8)
> table(true=tissue, cluster=hclusters)
cluster
true 1 2 3 4 5 6 7 8
cerebellum 0 0 31 0 0 2 5 0
colon 0 0 0 34 0 0 0 0
endometrium 15 0 0 0 0 0 0 0
hippocampus 0 12 19 0 0 0 0 0
kidney 37 0 0 0 0 2 0 0
liver 0 0 0 0 24 2 0 0
placenta 0 0 0 0 0 0 0 6
从上面两组计算方法我们可以看出来,除了个别情况外(例如endometrium和kidney),其余的簇中,基本上每簇代表一个组织。在某些情况下,一个组织有可能存在于两个簇中,这是因为选择的簇太多了。在聚类分析中,关于如何选择簇的个数也是一个很活跃的研究领域。
K-means
我们还可以使用kmeans()函数来进行k-means聚类。现在我们来演示一下如何使用这个函数,如下所示:
set.seed(1)
km <- kmeans(t(e[1:2,]), centers=7)
names(km)
mypar(1,2)
plot(e[1,], e[2,], col=as.fumeric(tissue), pch=16)
plot(e[1,], e[2,], col=km$cluster, pch=16)
结果如下所示:
> set.seed(1)
> km <- kmeans(t(e[1:2,]), centers=7)
> names(km)
[1] "cluster" "centers" "totss" "withinss" "tot.withinss"
[6] "betweenss" "size" "iter" "ifault"
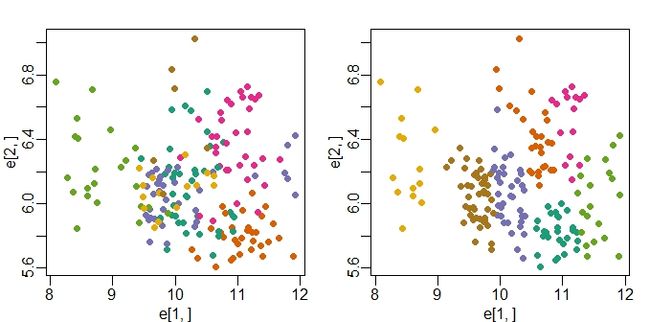
上图我们使用的是不同组织的前2个基因表达情况,其中颜色表示不同的组织。在在图中,颜色表示通过kmeans()函数计算的聚类结果。从下面的这个列表中我们就可以看到,这个聚类效果并不好:
table(true=tissue,cluster=km$cluster)
结果如下所示:
>
> table(true=tissue,cluster=km$cluster)
cluster
true 1 2 3 4 5 6 7
cerebellum 0 1 8 0 6 0 23
colon 2 11 2 15 4 0 0
endometrium 0 3 4 0 0 0 8
hippocampus 19 0 2 0 10 0 0
kidney 7 8 20 0 0 0 4
liver 0 0 0 0 0 18 8
placenta 0 4 0 0 0 0 2
这个结果不太好很有可能就是我们选择的这2个基因信息量不足以将不同组织区分开来,如果我们使用所有的基因进行kmeans计算,那么我们就能极大地改善聚类结果,现在我们使用MDS图来展示这个结果:
km <- kmeans(t(e), centers=7)
mds <- cmdscale(d)
mypar(1,2)
plot(mds[,1], mds[,2])
plot(mds[,1], mds[,2], col=km$cluster, pch=16)
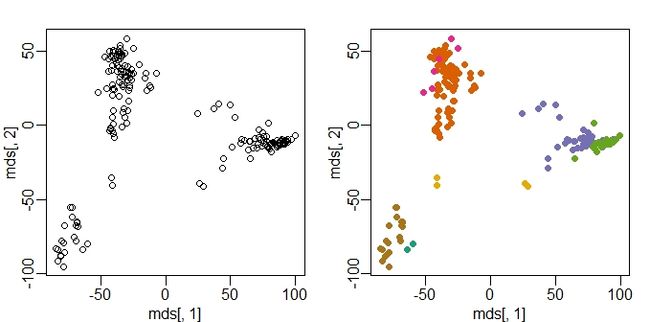
上图礣是使用前两个PC来绘制的聚类结果,右图是使用所有的基因来绘制的MDS图,使用颜色标明了不同的组织。
通过查看结果中的列表,我们就可以看到聚类的效果,如下所示:
table(true=tissue,cluster=km$cluster)
结果如下所示:
> table(true=tissue,cluster=km$cluster)
cluster
true 1 2 3 4 5 6 7
cerebellum 0 0 5 0 31 2 0
colon 0 34 0 0 0 0 0
endometrium 0 15 0 0 0 0 0
hippocampus 0 0 31 0 0 0 0
kidney 0 37 0 0 0 2 0
liver 2 0 0 0 0 0 24
placenta 0 0 0 6 0 0 0
热图
在遗传学文献中,热图的使用非常广泛。它们能够查看所有样本的不同基因的表达情况,有的时候还会在热图的上面或旁边添加上聚类后的树状图。现在我们来看一下如何创建热图,如下所示:
library(RColorBrewer)
hmcol <- colorRampPalette(brewer.pal(9, "GnBu"))(100)
library(genefilter)
rv <- rowVars(e)
# We will create heatmp using th e50 most variable genes and the function heatmap.2
idx <- order(-rv)[1:40]
现在我们使用gplots包中的heatmap.2了娄来绘制热图,并在热图的顶部添加组织信息,如下所示
library(gplots) ##Available from CRAN
cols <- palette(brewer.pal(8, "Dark2"))[as.fumeric(tissue)]
head(cbind(colnames(e),cols))
heatmap.2(e[idx,], labCol=tissue,
trace="none",
ColSideColors=cols,
col=hmcol)
结果如下所示:
> head(cbind(colnames(e),cols))
cols
[1,] "GSM11805.CEL.gz" "#1B9E77"
[2,] "GSM11814.CEL.gz" "#1B9E77"
[3,] "GSM11823.CEL.gz" "#1B9E77"
[4,] "GSM11830.CEL.gz" "#1B9E77"
[5,] "GSM12067.CEL.gz" "#1B9E77"
[6,] "GSM12075.CEL.gz" "#1B9E77"
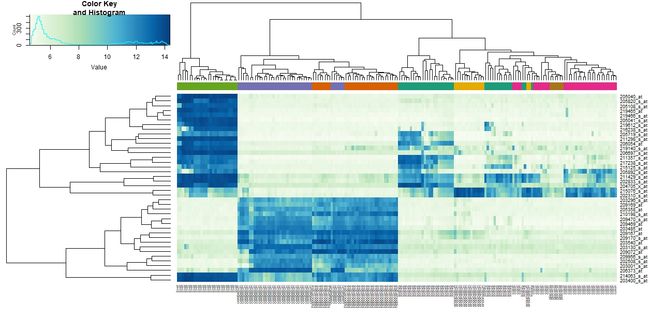
在热图中我们并没有使用组织信息,我们仅用了最显著的40个基因就发现了不同的组织。
练习
P374