如果只是做单个样本的sc-RNA-seq数据分析,并不能体会到Seurat的强大,因为Seurat天生为整合而生。
本教程展示的是两个pbmc数据(受刺激组和对照组)整合分析策略,执行整合分析,以便识别常见细胞类型以及比较分析。虽然本例只展示了两个数据集,但是本方法已经能够处理多个数据集了。
整个分析的目的:
- 识别两个数据集中都存在的细胞类型
- 在对照组和受刺激组均存在的细胞类型标记(cell type markers)
- 比较数据集,找出对刺激有反应的特殊细胞类型(cell-type)
数据准备
我已经下载好数据了,但是:
遇到的第一个问题就是,数据太大在windows上Rstudio连数据都读不了。谁叫我是服务器的男人呢,Windows读不了没关系啊,我到服务器上操作,生成rds在读到Rstudio里面。然后就遇到
scRNAseq.integrated <- RunUMAP(object = scRNAseq.integrated, reduction = "pca", dims = 1:30)
Error in RunUMAP.default(object = data.use, assay = assay, n.neighbors = n.neighbors, :
Cannot find UMAP, please install through pip (e.g. pip install umap-learn).
我明明已经装了umap-learn了呀,而且本地跑RunUMAP没问题,投递上去就不行。Google了半天,原来是conda的Python与R之间的调度不行,于是
library(reticulate)
use_python("pathto/personal_dir/zhouyunlai/software/conda/envs/scRNA/bin/python")
可以了。
library(Seurat)
library(cowplot)
ctrl.data <- read.table(file = "../data/immune_control_expression_matrix.txt.gz", sep = "\t")
stim.data <- read.table(file = "../data/immune_stimulated_expression_matrix.txt.gz", sep = "\t")
# Set up control object
ctrl <- CreateSeuratObject(counts = ctrl.data, project = "IMMUNE_CTRL", min.cells = 5)
ctrl$stim <- "CTRL"
ctrl <- subset(ctrl, subset = nFeature_RNA > 500)
ctrl <- NormalizeData(ctrl, verbose = FALSE)
ctrl <- FindVariableFeatures(ctrl, selection.method = "vst", nfeatures = 2000)
# Set up stimulated object
stim <- CreateSeuratObject(counts = stim.data, project = "IMMUNE_STIM", min.cells = 5)
stim$stim <- "STIM"
stim <- subset(stim, subset = nFeature_RNA > 500)
stim <- NormalizeData(stim, verbose = FALSE)
stim <- FindVariableFeatures(stim, selection.method = "vst", nfeatures = 2000)
在针对SeuratV3 的文章Comprehensive integration of single cell data中Anchors 是十分核心的概念。翻译成汉语叫做锚也就是基于CCA的一种数据比对(alignment)的方法。所以这两个函数亦需要看一下,以这样的方式来找到两个以致多个数据集的共有结构,这不是代替了之前的函数RunCCA()的应用场景了吗?
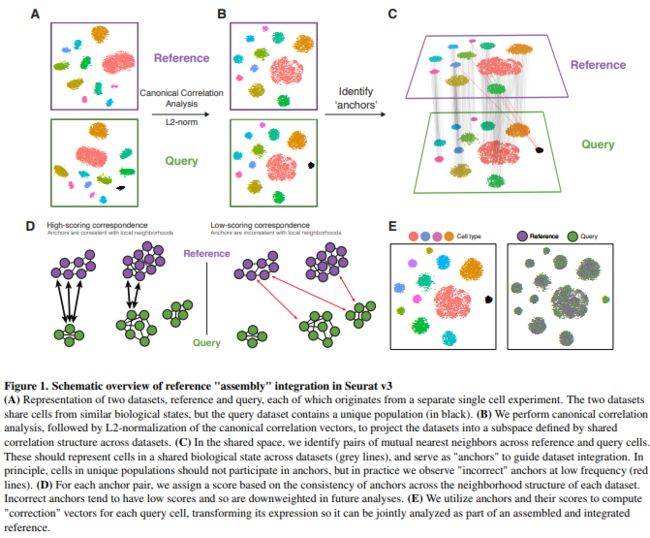
##Perform integration
?FindIntegrationAnchors
?IntegrateData
immune.anchors <- FindIntegrationAnchors(object.list = list(ctrl, stim), dims = 1:20)
immune.combined <- IntegrateData(anchorset = immune.anchors, dims = 1:20)
整合完之后,下面的操作就比较熟悉了,和单样本的思路一样。
#Perform an integrated analysis
DefaultAssay(immune.combined) <- "integrated"
# Run the standard workflow for visualization and clustering
immune.combined <- ScaleData(immune.combined, verbose = FALSE)
immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)
# t-SNE and Clustering
immune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:20)
immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:20)
immune.combined <- FindClusters(immune.combined, resolution = 0.5)
以上,都是我在服务上跑的,所以我要把他们读进来:
immune.combined<-readRDS("D:\\Users\\Administrator\\Desktop\\RStudio\\single_cell\\seurat_files_nbt\\seurat_files_nbt\\immune.combined_tutorial.rds")
> immune.combined
An object of class Seurat
16053 features across 13999 samples within 2 assays
Active assay: integrated (2000 features)
1 other assay present: RNA
2 dimensional reductions calculated: pca, umap
# Visualization
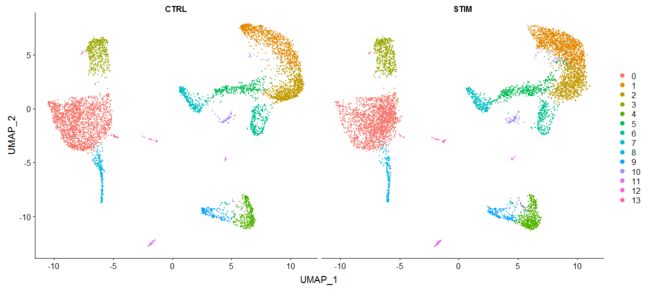
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE)
plot_grid(p1, p2)

可以用split.by 参数来分别展示两个数据:
DimPlot(immune.combined, reduction = "umap", split.by = "stim")
Identify conserved cell type markers
所谓保守的和高变的是对应的,也可以理解为两个数据集中一致的markers.FindConservedMarkers()函数对两个数据集执行差异检验,并使用MetaDE R包中的meta分析方法组合p值。例如,我们可以计算出在cluster 6 (NK细胞)中,无论刺激条件如何,都是保守标记的基因。但凡遇到差异分析的部分都会比较耗时。
#Identify conserved cell type markers
? FindConservedMarkers
DefaultAssay(immune.combined) <- "RNA"
nk.markers <- FindConservedMarkers(immune.combined, ident.1 = 7, grouping.var = "stim", verbose = FALSE)
head(nk.markers)
CTRL_p_val CTRL_avg_logFC CTRL_pct.1 CTRL_pct.2 CTRL_p_val_adj STIM_p_val STIM_avg_logFC STIM_pct.1 STIM_pct.2 STIM_p_val_adj max_pval minimump_p_val
GNLY 0 4.186117 0.943 0.046 0 0.000000e+00 4.033650 0.955 0.061 0.000000e+00 0.000000e+00 0
NKG7 0 3.164712 0.953 0.085 0 0.000000e+00 2.914724 0.952 0.082 0.000000e+00 0.000000e+00 0
GZMB 0 2.915692 0.839 0.044 0 0.000000e+00 3.142391 0.898 0.061 0.000000e+00 0.000000e+00 0
CLIC3 0 2.407695 0.601 0.024 0 0.000000e+00 2.470769 0.629 0.031 0.000000e+00 0.000000e+00 0
FGFBP2 0 2.241968 0.500 0.021 0 9.524349e-156 1.483922 0.259 0.016 1.338457e-151 9.524349e-156 0
CTSW 0 2.088278 0.537 0.030 0 0.000000e+00 2.196390 0.604 0.035 0.000000e+00 0.000000e+00 0
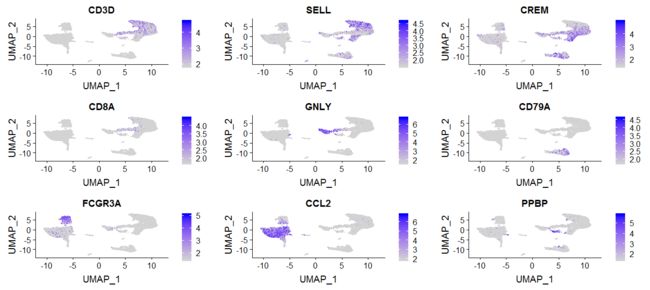
FeaturePlot(immune.combined, features = c("CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A",
"CCL2", "PPBP"), min.cutoff = "q9")
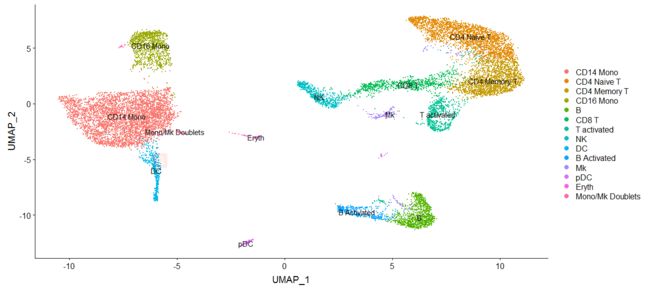
immune.combined <- RenameIdents(immune.combined, `0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T",
`3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "T activated", `7` = "NK", `8` = "DC", `9` = "B Activated",
`10` = "Mk", `11` = "pDC", `12` = "Eryth", `13` = "Mono/Mk Doublets")
DimPlot(immune.combined, label = TRUE)
Idents(immune.combined) <- factor(Idents(immune.combined), levels = c("Mono/Mk Doublets", "pDC",
"Eryth", "Mk", "DC", "CD14 Mono", "CD16 Mono", "B Activated", "B", "CD8 T", "NK", "T activated",
"CD4 Naive T", "CD4 Memory T"))
markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5",
"CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1",
"GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ")
DotPlot(immune.combined, features = rev(markers.to.plot), cols = c("blue", "red"), dot.scale = 8,
split.by = "stim") + RotatedAxis()
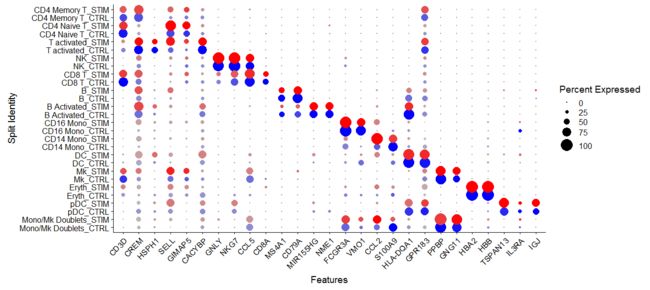
差异基因
在这里,我们取受刺激和受控制的原始T细胞和CD14单核细胞群的平均表达量,并生成散点图,突出显示对干扰素刺激有显著反应的基因。
#Identify differential expressed genes across conditions
t.cells <- subset(immune.combined, idents = "CD4 Naive T")
Idents(t.cells) <- "stim"
avg.t.cells <- log1p(AverageExpression(t.cells, verbose = FALSE)$RNA)
avg.t.cells$gene <- rownames(avg.t.cells)
cd14.mono <- subset(immune.combined, idents = "CD14 Mono")
Idents(cd14.mono) <- "stim"
avg.cd14.mono <- log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA)
avg.cd14.mono$gene <- rownames(avg.cd14.mono)
genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8")
p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) + geom_point() + ggtitle("CD4 Naive T Cells")
p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) + geom_point() + ggtitle("CD14 Monocytes")
p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)
plot_grid(p1, p2)
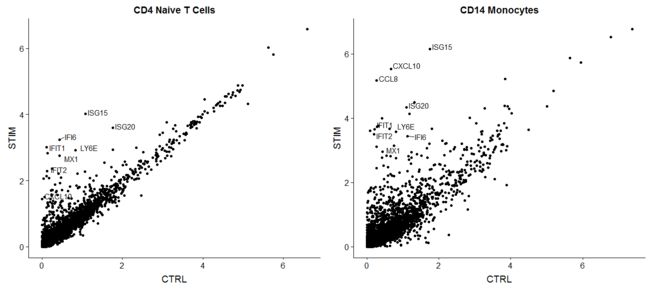
我们来用FindMarkers()看看这些基因是不是marker基因。
immune.combined$celltype.stim <- paste(Idents(immune.combined), immune.combined$stim, sep = "_")
immune.combined$celltype <- Idents(immune.combined)
Idents(immune.combined) <- "celltype.stim"
b.interferon.response <- FindMarkers(immune.combined, ident.1 = "B_STIM", ident.2 = "B_CTRL", verbose = FALSE)
head(b.interferon.response, n = 15)
p_val avg_logFC pct.1 pct.2 p_val_adj
ISG15 8.611499e-155 3.1934171 0.998 0.236 1.210174e-150
IFIT3 1.319470e-150 3.1195144 0.965 0.053 1.854251e-146
IFI6 4.716672e-148 2.9264004 0.964 0.078 6.628339e-144
ISG20 1.061563e-145 2.0390802 1.000 0.664 1.491814e-141
IFIT1 1.830963e-136 2.8706318 0.909 0.030 2.573053e-132
MX1 1.775606e-120 2.2540787 0.909 0.118 2.495259e-116
LY6E 2.824749e-116 2.1460522 0.896 0.153 3.969620e-112
TNFSF10 4.227184e-109 2.6372382 0.785 0.020 5.940461e-105
IFIT2 4.627440e-106 2.5102230 0.789 0.038 6.502941e-102
B2M 1.344345e-94 0.4193618 1.000 1.000 1.889208e-90
PLSCR1 5.170871e-94 1.9769476 0.794 0.113 7.266624e-90
IRF7 1.451494e-92 1.7994058 0.838 0.190 2.039785e-88
CXCL10 6.201621e-84 3.6906104 0.650 0.010 8.715138e-80
UBE2L6 1.324818e-81 1.4879509 0.854 0.301 1.861767e-77
PSMB9 1.098134e-76 1.1378896 0.940 0.571 1.543208e-72
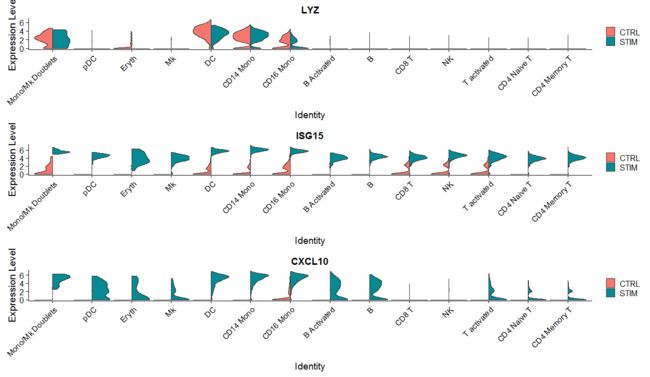
这里构造数据的过程值得玩味,然后绘制两样本的小提琴图,那么问题来了:两个以上数据集的小提琴图要如何绘制呢?
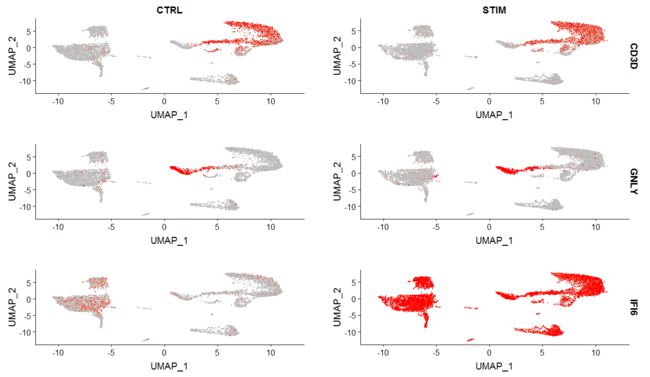
FeaturePlot(immune.combined, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3,
cols = c("grey", "red"))
plots <- VlnPlot(immune.combined, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim", group.by = "celltype",
pt.size = 0, combine = FALSE)
CombinePlots(plots = plots, ncol = 1)
Integrating stimulated vs. control PBMC datasets to learn cell-type specific responses
https://github.com/satijalab/seurat/issues/1020