蛋白质二级、三级结构预测
一.蛋白质结构预测
1.什么是蛋白质结构预测?
蛋白质是生命活动的基本单位,其结构决定了功能,对蛋白质结构的研究有助于对其功能的研究。上世纪50年代初,Anfinsen等人提出蛋白质的空间结构是由其一级结构决定。
蛋白质都是由20种不同的L型α氨基酸连接形成的多聚体,在形成蛋白质后,这些氨基酸又被称为残基。

蛋白质的分子结构可划分为四级,以描述其不同的方面:
• 蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。
• 蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。
• 蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。
• 蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
测定蛋白质序列比测定蛋白质结构容易得多,而蛋白质结构可以给出比序列多得多的关于其功能机制的信息。
蛋白质结构预测指的是:已知氨基酸序列,通过计算的手段预测蛋白质的二级结构和空间三维结构。
获得蛋白质 序列数据要比获得结构数据简单得多, DNA测序 技术的突飞猛进更使得可直接通过翻译、推导得 到大量的蛋白质序列. 而目前蛋白质结构数据库 PDB中所存储的蛋白质三维结构主要通过X 射线晶体衍射和核磁共振成像技术得到, 两种实验方法 均成本不菲, 且有各自的应用局限.
截止2016年 5 月, PDB 数据库中存储了11万余条蛋白质结构 数据, 而这只占UniProt中所有蛋白质序列数据的 1/600, 也就是说只有不到0.2%的蛋白质序列拥有 实验测定的三维结构。(来自2016年的物理学报文章)
2.蛋白质结果预测如何实现?
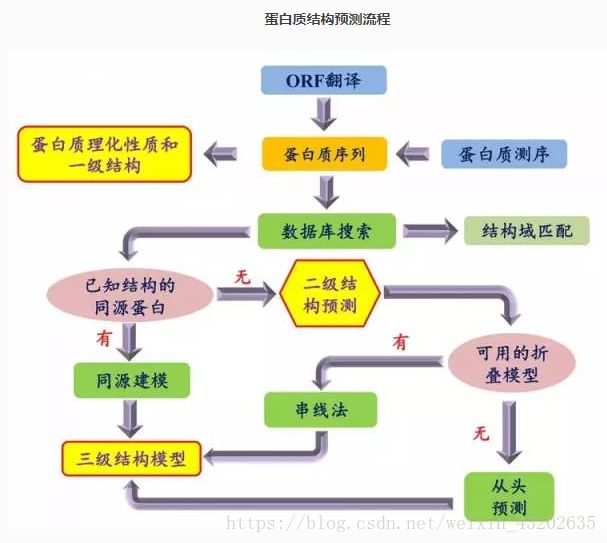
(1)基于模板的结构预测方法:同源建模+穿线方法
由于相似的蛋白质序列往往拥有相似的三维结构, 这就有了以PDB数据库中的已知结构为模板的同源建模方法, 它是迄今为止精度最高的一类结构预测方法.(来自2016年的物理学报文章)
而当PDB数据库中找不到与待预测蛋白质序列(下文中称为“目标 蛋白”或“目标序列”)具有显著序列相似性的蛋白质结构时, 此时通过穿线方法仍有可能找出与目标蛋白具有结构相似性的已知结构.
穿线方法——穿线方法实际上是通过某种策略将序列与结构进行比对, 评估将序列以各种匹配方式“安放”到各个三维结构上的“舒适”程度, 因此也被称为折叠辨识。
注:由于蛋白质结构 远比序列具有更高的保守性,毫无序列相似性的两个蛋白质也可能拥有相似的结构, 这是穿线法发挥作用的领域.
(2)无模板的结构预测方法——从头预测方法
从头预测方法不依赖于任何已知结构, 而是以第一性原理构建蛋白质折叠力场, 再通过相应的构象搜索方法搜寻目标蛋白的天然结构.
第一性原理是从头计算,不需要任何参数,只需要一些基本的物理常量,就可以得到体系基态的基本性质的原理。
无模板的结构预测方法与基于模板的预测方法的关键区别在于: 它们并不依赖于任何一个完整的结构模 板, 不要求片段所在模板与目标蛋白有任何同源性 或结构相似性, 这让它们具有更大的随机性和自由 度, 便于模拟已知结构中不存在的全新结构.
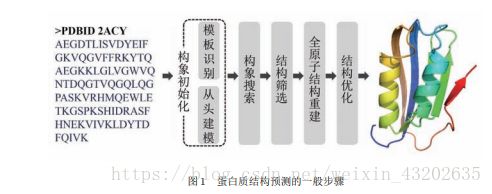
㈠构象初始化
“基于模板” 和“无模板” 两类结构预测方法的关键区别——就在于所采取的构象初始化方法不同:
基于模板的预测方法通过搜索识别与目标蛋白具有同源性或结构相似性的已知结构作为模板而获得初始化构象;(提高序列比对方法的效率、精度是促进同源建模方法发展的关键.)
无模板的预测方法则通常以小的结构片段为起点从头构建初始构象. 许多无模板的结构预测方法所采用的结构片段装配就是以预测得到的二级结构、主链二面角以及溶剂可及性为依据, 从 PDB 数据库中截取 一系列小的结构片段, 通过片段装配得到目标蛋白完整的初始构象.
㈡构象搜索
获得初始化构象之后, 通常还需要进行构象搜索, 即在一定力场的指导下,采用某种搜索策略不断改变分子构象, 以搜寻更接近天然的构象.
注:α-氨基酸由一个所有氨基酸类型中都含有的共同部分(形成蛋白质的主链)和一个对每一类氨基酸都不同的侧链所组成。侧链决定了20种α-氨基酸的化学性质。
力场是一种矢量场,其中与每一点相关的矢量均可用一个力来度量。
进行构象搜索必须要有一个能描述蛋白质构象能量景貌的力场, 目标蛋白 的任意一个构象都对应能量景貌上的一个点,
对于设计精良的力场来说,目标蛋白的天然构象应当位于能量景貌的最低点.
力场确定了蛋白质各个构象的能量值, 接着需要一定的方法来搜索低能量的构象.
基于物理的力场从第一性原理出发,物理意义明确,但精度不够,实际应用效果往往并不理想.
基于知识的力场从已知结构归纳提取特征,易于构建且性能表现不俗,但却规避了对力场物理本质的探索.
搜索方法:
分子动力学模拟——通过解牛顿运动方程来搜索构象, 计算量非常巨大
基于蒙特卡罗模拟的构象搜索——构象是在事先设计好的 各种随机变动中不断变化折叠,变动的尺度、幅度以及各种变动的出现频率都可 以根据需要进行设置和调整,同时为了跨越能量壁垒,在构象搜索过程中采用模拟退火、副本交换等策略, 同时还配合使用多个初始化构象,从能量景貌的不同位置开始构象搜索.
㈢结构筛选
模拟过程中通常会不断输出一些能量较低的中间结构以供后续筛选. 结构筛选其实与结构评估对应着一个相同的问题, 即如何正确区分不同质量 (与天然结构具有不同差异程度) 的预测结构.
构象搜索后得到的一般只是蛋白质的简化结构,发展优秀的结构筛选及评估方法是蛋白质结构预测中极为重要的研究方向。(CASP竞赛保存有大量预测结构)
筛选方法——利用更为复杂、精细的打分函数 (score function) 来筛选结构.
㈣全原子结构重建
由于目前的结构预测方法普遍都采用蛋白质 简化模型进行构象搜索,通过上述步骤得到的也 只是一个或多个简化结构, 接下来需要在简化结 构的基础上重建起全原子结构. 对于不同的简化 模型, 全原子重建的过程也不一样.方法有Cα-原子加 “虚拟侧链中心” 式的简化模型。
㈤结构优化
虽然通过前面的步骤已经获得了目标蛋白完 整的预测结构, 但由于所用力场、构象搜索方法以 及全原子重建方法自身的问题和缺陷, 该结构的 质量 (尤其是局部结构质量) 往往存在较大的优化空间.
有的结构预测方法 会在全原子结构重建的同时, 分步骤进行结构优化。结构优化过程的主要目的则是要在整体拓扑结构变动不大的情况下尽可能改善其局部结构细节. 最好的情况是使目标蛋白的整体拓扑结构以及局部结构细节均得到优化, 目前这仍是一个极具挑战的任务.
方法有:FG-MD ——基于分子动力 学模拟的结构优化方法,同时从不同结构模板和 小的结构片段中搜集空间限制信息来指导结构优化.
ModRefiner——基于蒙特卡罗模拟的结构优化方法,同时具备全原子结构重建的功 能, 其结构重建和结构优化的功能相辅相成.
注:自第七届起, 还专门设置了模型优化 (model refinement) 项目, 以评估蛋白质结构优化方面的发展情况。
3.现有的算法有哪些?效果如何?是否还有提升空间?
(1)基于模板的结构预测方法
SWISS-MODEL(基于同源建模)
Modeller(基于同源建模)
I-TASSER(基于穿线法)
(2)无模板的结构预测方法
无模板结构预测方法的发展不仅受实际应用的驱动 (并不是所有目标蛋白都能在结构数 据库中找到满意的模板), 更受到蛋白质折叠密码这一基本科学问题的推动.
Rosetta
QUARK
SCRATCH
注:由于计算量巨大、力场精度不够等原因, 目前无模板的结构预测方法还只能应对尺寸相对较小(<150 残基) 的目标蛋白.
4.国际蛋白质结构预测技术评估大赛(CASP)
在计算生物学领域已久负盛名, 常被誉为蛋白质结构预测的奥林匹克竞赛.
(1)比赛
所有目标蛋白将被划分成基于模板 (TBM) 和无模板 (FM) 两类. 所有参赛方法也被归为人工组 和自动组两类,人工组意味着综合了计算机预测 和人工干预,自动组则纯粹依赖计算机预测.
除了蛋白质三维结构预测, CASP 大赛还包括 对蛋白质结构其他方面的一些预测方法的评估, 如 残基接触预测、无序区域预测、结构质量评估、结构优化等.
(2)进展
二十余年来, CASP 竞赛全方位见证了蛋白质结构预测领域的发展. 在最初几届竞赛中,基于模板的预测方法得到的预测结构往往比模板本身更加偏离目标蛋白天然结构, 而在最近数届竞赛中, 许多目标蛋白的最佳预测结构都显著优于模板结构, 这主要得益于多模板综合建模的发展以 及 (基于知识的) 力场精度的提高. 无模板的结构 预测进步同样也很大, 目前对于 100 个左右残基长度的无模板类目标蛋白, 不少预测方法能够给 拓扑结构基本正确 (RMSD 约 4—10 Å) 的预测结 果, 少数例子中甚至能获得 RMSD 接近于 2 Å 的 预测结构.
(3)挑战
较大尺寸的蛋白质(>150残基)仍是无模板结构预测的最大挑战, 目前为止还 很难预测得到具有较高应用价值的结构. 促进无模板结构预测的发展将是 CASP 竞赛今后最重要的目标和着力方向.(CASP ROLL 竞 赛)
2016年的情况——目前的序列比对和穿线算法对目标蛋白远程同源模板的识别能力有限, 构象搜索力场的设计远不够精确, 整体拓扑结构与局部结构 细节难以同时获得优化, 实验难以测定的膜蛋白结构同样也是结构预测的软肋, 蛋白质复合体结构 (四级结构) 的预测仍困难重重等.
目前绝大部分的预测方法都过分依赖于已知的蛋白质结构信息, 即便是无模板 的结构预测方法, 几乎也无一幸免. 减少对已知结构的依赖, 强化对本质规律的研究, 这是未来蛋白 质结构预测寻求理论突破的必由之径.
二.蛋白质三级结构预测
目前蛋白三维结构模拟的依据是基于以下两点:
(1)在进化过程中,蛋白质的结构比序列更加保守;
(2)有证据表明,现在已知的蛋白质折叠方式是有限的,数量约1 000-10 000。
㈠构象初始化
“基于模板” 和“无模板” 两类结构预测方法的关键区别——就在于所采取的构象初始化方法不同:
基于模板的预测方法通过搜索识别与目标蛋白具有同源性或结构相似性的已知结构作为模板而获得初始化构象;(提高序列比对方法的效率、精度是促进同源建模方法发展的关键.)
无模板的预测方法则通常以小的结构片段为起点从头构建初始构象.许多无模板的结构预测方法所采用的结构片段装配就是以预测得到的二级结构、主链二面角以及溶剂可及性为依据,从 PDB 数据库中截取 一系列小的结构片段,通过片段装配得到目标蛋白完整的初始构象.
㈡构象搜索
获得初始化构象之后, 通常还需要进行构象搜索, 即在一定力场的指导下,采用某种搜索策略不断改变分子构象, 以搜寻更接近天然的构象.
进行构象搜索必须要有一个能描述蛋白质构象能量景貌的力场, 目标蛋白的任意一个构象都对应能量景貌上的一个点, 对于设计精良的力场来说,目标蛋白的天然构象应当位于能量景貌的最低点.力场确定了蛋白质各个构象的能量值,接着需要一定的方法来搜索低能量的构象.
基于物理的力场从第一性原理出发,物理意义明确,但精度不够,实际应用效果往往并不理想.
基于知识的力场从已知结构归纳提取特征,易于构建且性能表现不俗,但却规避了对力场物理本质的探索.
搜索方法:分子动力学模拟——通过解牛顿运动方程来搜索构象, 计算量非常巨大
基于蒙特卡罗模拟的构象搜索——构象是在事先设计好的 各种随机变动中不断变化折叠,变动的尺度、幅度以及各种变动的出现频率都可 以根据需要进行设置和调整,同时为了跨越能量壁垒,会在构象搜索过程中采用模拟退火、副本交换等策略, 同时还配合使用多个初始化构象,从能量景貌的不同位置开始构象搜索.
㈢结构筛选
模拟过程中通常会不断输出一些能量较低的中间结构以供后续筛选. 结构筛选其实与结构评估对应着一个相同的问题, 即如何正确区分不同质量 (与天然结构具有不同差异程度) 的预测结构.
构象搜索后得到的一般只是蛋白质的简化结构,发展优秀的结构筛选及评估方法是蛋白质结构预测中极为重要的研究方向。(CASP竞赛保存有大量预测结构)
筛选方法——利用更为复杂、精细的打分函数 (score function) 来筛选结构.
㈣全原子结构重建
由于目前的结构预测方法普遍都采用蛋白质简化模型进行构象搜索,通过上述步骤得到的也 只是一个或多个简化结构, 接下来需要在简化结构的基础上重建起全原子结构. 对于不同的简化模型, 全原子重建的过程也不一样.方法有Cα-原子加 “虚拟侧链中心” 式的简化模型。
㈤结构优化
虽然通过前面的步骤已经获得了目标蛋白完整的预测结构, 但由于所用力场、构象搜索方法以 及全原子重建方法自身的问题和缺陷, 该结构的质量 (尤其是局部结构质量) 往往存在较大的优化空间.
有的结构预测方法 会在全原子结构重建的同时, 分步骤进行结构优化。结构优化过程的主要目的则是要在整体拓扑结构变动不大的情况下尽可能改善其局部结构细节. 最好的情况是使目标蛋白的整体拓扑结构以及局部结构细节均得到优化, 目前这仍是一个极具挑战的任务.
方法有:FG-MD ——基于分子动力学模拟的结构优化方法,同时从不同结构模板和 小的结构片段中搜集空间限制信息来指导结构优化.
ModRefiner——基于蒙特卡罗模拟的结构优化方法,同时具备全原子结构重建的功 能, 其结构重建和结构优化的功能相辅相成.
三.蛋白质二级结构预测
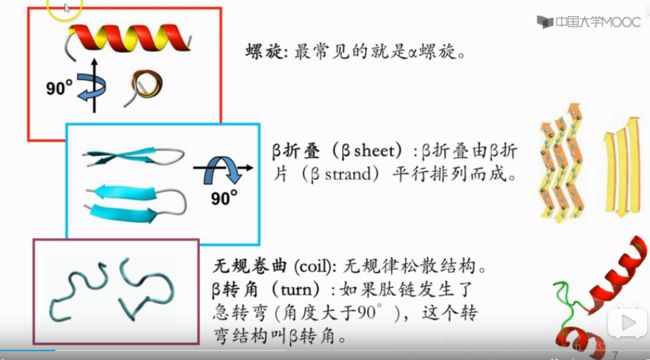
图1 蛋白质主要二级结构
蛋白质二级结构是氨基 酸残基在蛋白质多肽链中的局部空间构象,其具有 8 种类型,
分别是 α-螺旋(H)、β-桥(B)、折叠(E)、螺旋-3(G)、 螺旋-5(I)、转角(T)、卷曲(S)和环(L)。
把八类二级结构粗略地归类为螺旋、折叠和卷曲三类。
每个二级结构 类型都是由其所在蛋白质链中氨基酸残基间的局部和长程相 互作用共同决定的。蛋白质二级结构预测任务就是把一个由20 种氨基酸 A、C、D、E、F、G、H、I、K、L、M、N、P、 Q、R、S、T、V、W 和 Y 组成的氨基酸序列映射为对应的二级结构序列。
蛋白质二级结构预测分两个步骤:
首先,对数据集中的蛋白质序列进行特征提取,并构造训练集测试集;
然后,构建预测系统模型,用训练集对模型进行训练,优化模型参数,再用测试集测试模型的效果。
蛋白质序列的特征提取常用方法有2种:氨基酸组分(AAC)方法和伪氨基酸组分(PseAA)方法。
AAC仅仅描述了蛋白质序列中各氨基酸的含量百分比,丢失了各残基之间的顺序信息。
PseAA是在AAc的基础上,加入了一些能够表示残基之间顺序信息的特征。
蛋白质二级结构预测的方法有很多,如模糊聚类、贝叶斯分类、信息差、逻辑回归、决策树、人工神经网络(ANN)、马尔科夫模型、支持向量机(sVM)、K近邻、粗糙集等,其中,ANN、sVM等方法均取得了很好的预测精度。
1.蛋白质序列表示
蛋白质序列可表示为 P = R1R2R3…RN
其中Ri表示第i个氨基酸残基,这些残基属于20种氨基酸中的一种,P为长度为N的字符串。直接将P做输入来进行训练,同源性低的蛋白质会得到很差的结果。因此需要对P进行特征提取。
(1)ACC方法
用一个20维向量表示蛋白质:
X = [X1,X2,…X20]
其中Xi表示第i种氨基酸在序列P中出现的次数。
该方法提取的特征只能反映20种基本氨基酸的出现频率,丢失了各个残基之间的排列顺序信息。
(2)PseAA方法
用一个20+M维向量表示蛋白质:
X = [X1,X2,…X20,…,X20+M]
其中20维向量表示20种氨基酸残基的出现次数,另外M维向量表示蛋白质的近似熵、亲水疏水信息等特征。
2.基于深度学习的预测方法
采用人工神经网络建立预测模型:
包括一个输入层、一个输出层和K个隐含层。
输入层是一个20+M维的蛋白质特征向量;
输出层是一个4维向量,表示蛋白质所属类别。???????????????
训练阶段:
对于每个输入蛋白质,输出向量中对应的分类位置设为1,其余3个分量设为0;
测试阶段:
对于输入蛋白质,输出向量中最大分量所对应的类别作为该蛋白质的预测分类。
四.当前的研究水平
1.二级结构预测
已有大量有关根据序列预测蛋白质二级结构的文献资料,这些资料可大致分为二类: 一是有关根据单一序列预测二级结构; 二是有关根据多序列列线预测二级结构。
直到最近为止,二级结构预测才不被认为具有很高的随机性。大多数预测算法均是依据单一序列。即使是最著名的一些算法(如Chou-Fasman算法和GOR算法)也只有约60%的预测准确率,而对于一些特定的结构,如那些富含β-折叠片的结构,这些算法难以预测成功。预测失败的原因主要是单一序列所提供的信息只是残基的顺序而没有其空间分布的信息。
两个方面的研究进展改变了这一状况:一是认识到多序列列线可被用于改进预测能力。多序列列线可被视为诱变遗传学试验中的自然突变状况,其对序列上单一位点变异的分析的确提供了该位点在蛋白质三级结构中的信息;二是神经网络已开始被用于根据序列预测结构。
目前已有这样一个共识,即在有大量、高质量的多序列列线结果的情况下,蛋白质二级结构的预测将非常准确–通常准确率比以单一序列预测提高10%。
一些文献表明,一些程序**(诸如PHD)预测的准确率达到了目前最高水平**。 PHD (http://www.embl-heidelberg.de/predictprotein/predictprotein.html) 提供了从二级结构预测到折叠(fold)识别等一系列功能。
PHD被认为是二级结构预测的标准。
总的来说,二级结构预测仍是未能完全解决的问题,一般对于α螺旋预测精度较好,对β折叠差些,而对除α螺旋和β折叠等之外的无规则二级结构则效果很差。据报道,对于最佳实例的预测,nnpredict(PHD里的预测算法)的准确率超过了65%。

![]()



2.三级结构预测
根据序列或多序列列线预测蛋白质二级结构的技术已相对比较成熟,但三级结构的预测则相当困难。
往往对于三级结构预测,只能通过与已知结构蛋白序列同源性比对来完成。这一方法已是目前进行三级结构预测的最准确方法。但是这一方法并不总是奏效,因为大约有80%的已知蛋白质序列找不到与之相似的已知结构的蛋白质序列。
比对数据库中已知结构的序列是预测未知序列三级结构的主要方法。多种途径可进行以上这种比对。最容易是使用BLASTP程序比对NRL-3D或SCOP数据库中的序列。如果发现超过100个碱基长度且有远高于40%序列相同率的匹配序列,则未知序列蛋白与该匹配序列蛋白将有非常相似的结构。
在这种情况下,同源性建模(homology modeling)在预测该未知蛋白精细结构方面会发挥非常大的作用。在序列相同率为25%~40%时,两条蛋白质将具有相同的折叠,但这时同源性建模将变得更加困难和不准确。
蛋白质三维结构预测服务通过因特网向公众免费开放(同源建模):
瑞士生物信息研究所 SWISS-MODEL
丹麦技术大学生物序列分析中心 CPHmodels
比利时拿摩大学 ESyPred3D
英国癌症研究中心 3DJigsaw

